Date: Fri, 19 Aug 2016 12:56:24 +0530 (IST)
Dear AMBER users,
I had performed a Targeted MD simulation for my mutant protein.This is
my input file -
targeted MD . 20*c
&cntrl
imin = 0, nstlim = 30000, dt = 0.0007,
ntx = 1, ntt = 0,
ntc = 2, ntf = 2, ntb = 0,
cut = 999.0,
ntpr = 10, ntwx = 10, ntwr = 10,
igb = 1, irest=0, nmropt=1,tempi=293.0,
itgtmd=1, tgtrmsd=1 tgtmdfrc=1.00,
tgtfitmask=":1-520.CA,N,C,O",
tgtrmsmask=":1-520 & !.H=",
/
&wt
TYPE='TGTRMSD', istep1 =1, istep2 = 30000,
value1 = 30.6, value2 = 1.0,
/
&wt
type="END",
/
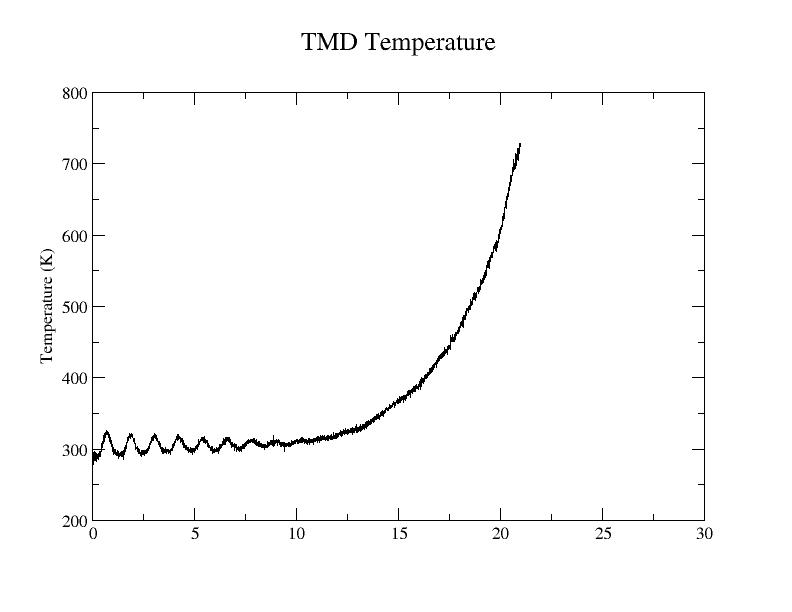
I have set the temperature of the system at 293 K. But as TMD progress the
temperature of the system shoots-up to 800K.
Is it normal?
Will the protein not get denatured at 800K?
Is there any way to maintain temp constant?
Why is there a temperature shoot-up?
I have attached the graph of temperature along TMD with this mail.
Thanking u in advance,
Aravind R
======================================================================
Aravind R
Research Associate
Institute of Bioinformatics and Applied Biotechnology
Bangalore-560100
ph-(+91)888-46-11-500
======================================================================
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: temp.jpg)
