Date: Mon, 15 Aug 2016 15:08:25 +0000
Good morning,
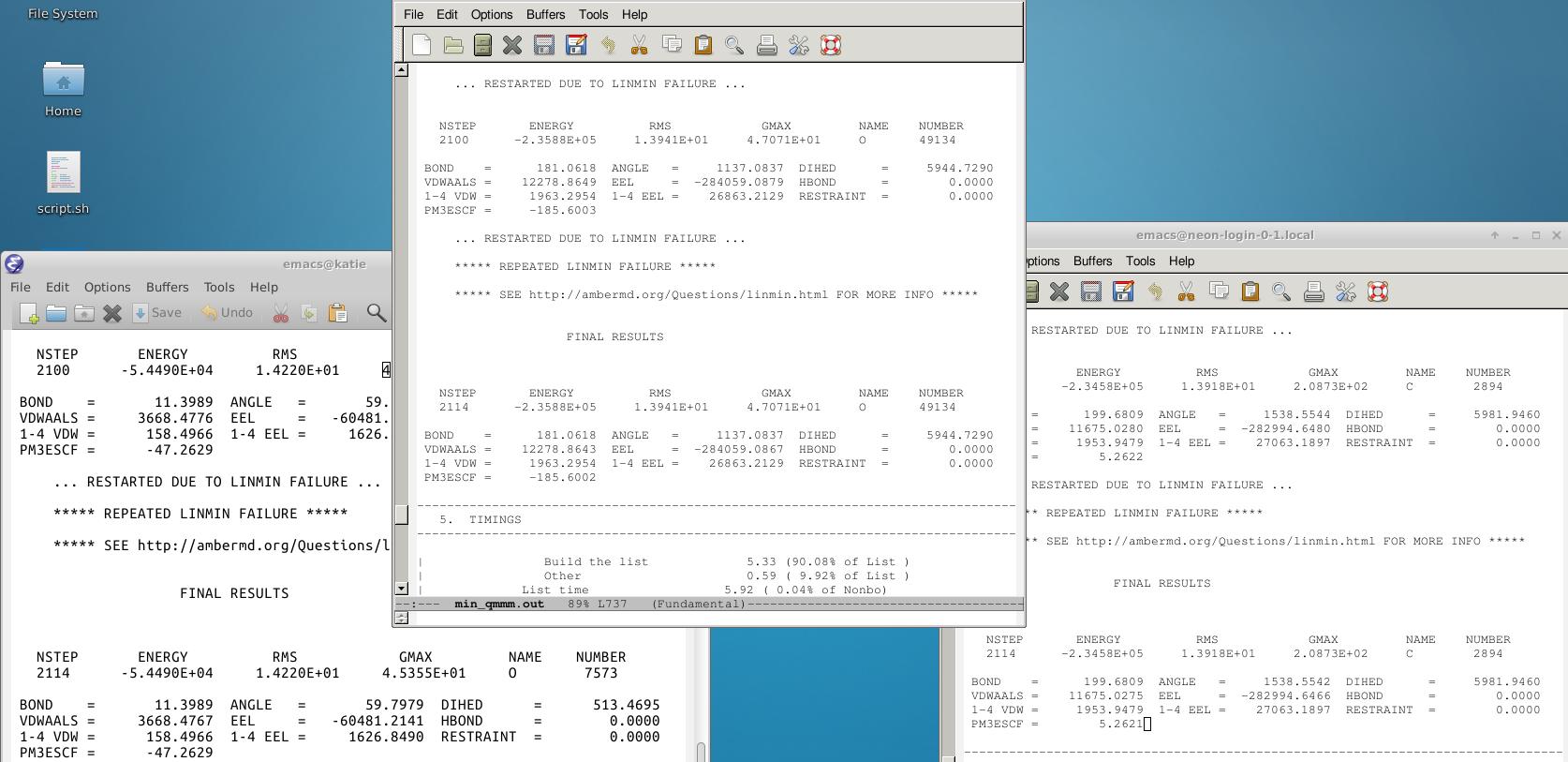
I am trying to run a QM/MM calculation on some proteins using Amber16. I did the NMA tutorial and I based my calculation off of that. After I ran the first protein (output on the left in the image at the bottom) I thought the QM region of the structure was optimized due to the fact that the output says final results. I was excited and moved onto more calculations (middle and right image). However each calculation stops at NSTEP 2114. I don't think this is a coincidence. I think I need to restart the calculation from the rst file but I am not sure how to do that. Is that what I should do since I don't think the QM region is optimized? Below is the input file. I used the same parameters for all 3 calculations but the qmmask value differs for all 3. I also put each protein into a box of water molecules using tleap.
Thank you in advance! - Katie
Initial min of our structure QMMM
&cntrl
imin=1, maxcyc=5000, ncyc=2000,
cut=20.0, ntb=1, ntc=2, ntf=2,
ifqnt=1
/
&qmmm
qmmask= ':7,70,181,183,511'
qmcharge= -1,
qm_theory='PM3',
qmshake=1,
qm_ewald=1, qm_pme=1
/
[cid:f63808ed-e6d3-48cc-b268-813ea3db799b]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: pastedImage.png)
