Date: Mon, 4 Jul 2016 17:02:54 -0300
Dear Fabrizio, good afternoon.
I needed for a study I'm doing here to create a modified residue
Lysine carboxylated attached to the enzyme active site.
That is, in my case had a COO waste is not standard in the active site.
When I see it looks similar to what you intend to do.
First I did the following.
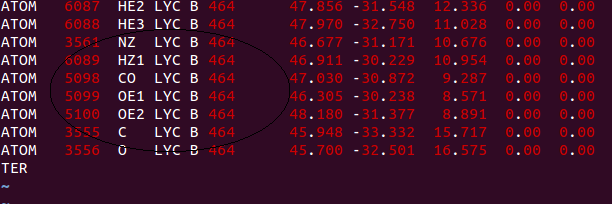
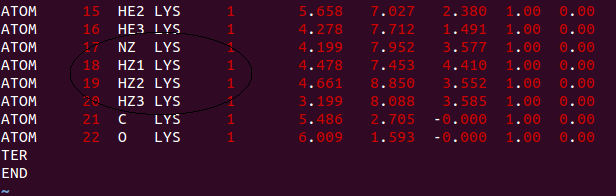
I - I created a .pdb file with the waste that needed to modify (in my case
the N atom appeared in the file attached to it three H atoms, as shown
below;
.
.
.
.[image: Imagem intercalada 1]
[image: Imagem intercalada 2]
II - replaces this place for the group needed to put (COO), and used
the following tutorial as a reference:
http://ambermd.org/tutorials/advanced/tutorial1_adv/
III - I had to parameterize Some constant values of strength and angle,
and got
charges for online server RED
IV - I could create .frcmod and .mol2 files to this modified amino acid.
You will need to do something, because when I use MCPB.py had to provide
these input files (which for the modified amino acid residue
They are understood in xleap as non-standard waste).
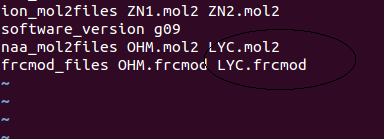
See below my .in file for MCPB.py
.
.
.
[image: Imagem intercalada 3]
Note that contains files related to what I am commenting.
In my case, after using MCPB.py and get the files .mol2 the program
LY1.mol2 created another file besides the other for other waste
amino acids of the metal coordination sphere which I am using.
You'll have to create a non-standard modified residue, as this modified
residue should appear on the related site in your .pdb file protein
all and has to be recognized with the parameters AMBER force field
in xleap.
I hope I have not acid very confusing to understand.
Best regards
Marcelo A. Chagas
Marcelo Andrade Chagas, MSc
(PhD student)
Laboratório de Química Computacional e Modelagem Molecular - LQC-MM
* http://lqcmm.qui.ufmg.br/
Departamento de Química da Universidade Federal de Minas Gerais - UFMG
Tel:(31)3409-5776
2016-07-04 15:49 GMT-03:00 Fabrício Bracht <fabracht1.gmail.com>:
> Hello again.
> I was able to execute all steps of MCPB.py and generate the tleap.in
> script, but there seems to be a problem with the modified Histidine
> residue. There are no parameters for the bond between the carbonyl carbon
> atom and the nitrogen of residue number 2 on the protein (there aren't
> parameters for angles and dihedrals as well). Here is the tleap warning:
>
> Building bond parameters.
> Could not find bond parameter for: c1 - N
> Building angle parameters.
> Could not find angle parameter: o - c1 - N
> Could not find angle parameter: c1 - N - H
> Could not find angle parameter: c1 - N - CX
> Could not find angle parameter: c3 - c1 - N
> Building proper torsion parameters.
> ** No torsion terms for o-c1-N-H
> ** No torsion terms for o-c1-N-CX
> ** No torsion terms for c3-c1-N-H
> ** No torsion terms for c3-c1-N-CX
>
> c1 (lower case c) refers to the histidine residue in question that was
> originally considered a ligand and N (upper case N) is probably the
> nitrogen atom of the residue to which this histidine is bonded to.
> I'm not sure what CX refers to though.
> There are some other problems also. The mol2 file for the Histidine has 0
> charges for all heavy atoms.
>
> Any help here would be great.
> Thank you
> Fabrício
>
> 2016-07-01 20:29 GMT-03:00 Fabrício Bracht <fabracht1.gmail.com>:
>
> > Hi Pengfei and Marcelo. Now I get it. Plus, I wrote to the gaussian guys
> > to ask why the large_mk.com calculation was terminating with an error
> > "Error termination via Lnk1e in /home/fabricio/g09/l602.exe at Mon". The
> > answer was to add a line with the name of the file into which the ESP
> > charges will be written. It is important to add a blank line between the
> > Copper MKradius value and to add two blank lines after the filename (I´ve
> > tested a bit to see if that really mattered).
> > Things seem to be getting on the right track now.
> > Thanks
> > Fabrício
> >
> > 2016-06-30 13:51 GMT-03:00 Pengfei Li <ambermailpengfei.gmail.com>:
> >
> >> Hi Fabricio,
> >>
> >> I guess Marcelo's suggestion is about performing the partial
> optimization
> >> with only the external part being optimized but the central part being
> >> frozen.
> >>
> >> In Gaussian a frozen symbol -1 or optimize symbol 0 follows the element
> >> symbol and aheads the atomic coordinates is used to freeze/free certain
> >> atom(s) during the optimization. For example:
> >>
> >> C -1 0.000 0.000 0.000
> >> H 0 1.000 0.000 0.000
> >>
> >> means only optimize the position of H but freeze the position of C
> during
> >> the optimization (also don’t forget to use opt keyword in the Gaussian
> >> input file).
> >>
> >> Is that right? Marcelo.
> >>
> >> Kind regards,
> >> Pengfei
> >>
> >> > On Jun 29, 2016, at 12:57 PM, Marcelo Andrade Chagas <
> >> andrade.mchagas.gmail.com> wrote:
> >> >
> >> > Dear Fabrizio, good afternoon.
> >> >
> >> > I'm also using MCPB.py program to study Bimetallic enzyme systems.
> >> >
> >> > Today even managed to complete the steps until you reach the creation
> of
> >> > topology files and inicais speeds.
> >> >
> >> > As for your question, the principle by which I understand is the
> >> following:
> >> >
> >> > the most active site model you will make an optimization and then
> >> perform
> >> > a charge calculation. Because the key words (IOPS) used in the input
> if
> >> you
> >> > open
> >> > the output file .log corresponding gaussian in the / Initial
> Parameters
> >> > you will see that during the calculation of the sitema is frozen and
> >> > optimization is performed
> >> > only on the most external part of the system under study.
> >> >
> >> > This is Pengfei?
> >> >
> >> > Best regards
> >> >
> >> > Marcelo Andrade Chagas, MSc
> >> > (PhD student)
> >> > Laboratório de Química Computacional e Modelagem Molecular - LQC-MM
> >> > * http://lqcmm.qui.ufmg.br/
> >> > Departamento de Química da Universidade Federal de Minas Gerais - UFMG
> >> > Tel:(31)3409-5776
> >> >
> >> > 2016-06-29 13:32 GMT-03:00 Fabrício Bracht <fabracht1.gmail.com>:
> >> >
> >> >> Hi Pengfei.
> >> >> I have a question regarding the gaussian calculation of the large
> >> model.
> >> >> From the input file, I can see that no geometry optimization is
> >> performed
> >> >> on this model. I encountered convergence problems with this step. I
> am
> >> >> guessing that, since the geometry of the complex obtained directly
> >> from the
> >> >> pdb is not that great, the SCF routine has problems with convergence
> >> (hence
> >> >> the XQC flag). Is that correct?
> >> >> But even so, the large model gaussian calculation terminates with an
> >> error.
> >> >> Is there something else I could do to fix this?
> >> >>
> >> >> Thanks
> >> >> Fabrício
> >> >>
> >> >> 2016-06-28 11:33 GMT-03:00 Pengfei Li <ambermailpengfei.gmail.com>:
> >> >>
> >> >>> Hi Fabricio,
> >> >>>
> >> >>> I have modified MCPB.py code to make it can work for your case. And
> I
> >> >> have
> >> >>> sent an email to your email address about that. Hope it helps.
> >> >>>
> >> >>> Kind regards,
> >> >>> Pengfei
> >> >>>
> >> >>>> On Jun 27, 2016, at 3:56 PM, Fabrício Bracht <fabracht1.gmail.com>
> >> >>> wrote:
> >> >>>>
> >> >>>> Hello. I've given up on using MCPB.py, and am trying to use MCPB
> >> >> instead.
> >> >>>> I need to create a Histidine residue that has a methyl group bonded
> >> to
> >> >>> the
> >> >>>> epsilon nitrogen instead of the hydrogen that would be there.
> >> >>>> So far I've tried to introduce a terminal CH3 with the command:
> >> >>>>
> >> >>>> addFragment terminal/CH3 bd /NAME/CLR/HD1-1/.NE2 ag
> >> >> /NAME/CLR/HD1-1/.CD2
> >> >>> tr
> >> >>>> /NAME/CLR/HD1-1/.CE1 165.00
> >> >>>>
> >> >>>> This works fine, but the HE2 is still there. There is no command
> >> listed
> >> >>> on
> >> >>>> the manual to remove atoms. I could, change the HIE to a HID and
> >> >> transfer
> >> >>>> the hydrogen to the other nitrogen atom, but the other nitrogen is
> >> >> bonded
> >> >>>> to the metal ion.
> >> >>>> Can I replace atoms or even remove them in MCPB?
> >> >>>>
> >> >>>> Thank you
> >> >>>> Fabrício
> >> >>>> _______________________________________________
> >> >>>> AMBER mailing list
> >> >>>> AMBER.ambermd.org
> >> >>>> http://lists.ambermd.org/mailman/listinfo/amber
> >> >>>
> >> >>>
> >> >>> _______________________________________________
> >> >>> AMBER mailing list
> >> >>> AMBER.ambermd.org
> >> >>> http://lists.ambermd.org/mailman/listinfo/amber
> >> >>>
> >> >> _______________________________________________
> >> >> AMBER mailing list
> >> >> AMBER.ambermd.org
> >> >> http://lists.ambermd.org/mailman/listinfo/amber
> >> >>
> >> > _______________________________________________
> >> > AMBER mailing list
> >> > AMBER.ambermd.org
> >> > http://lists.ambermd.org/mailman/listinfo/amber
> >>
> >>
> >> _______________________________________________
> >> AMBER mailing list
> >> AMBER.ambermd.org
> >> http://lists.ambermd.org/mailman/listinfo/amber
> >>
> >
> >
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
(image/png attachment: 02-image.png)
(image/png attachment: 03-image.png)
