Date: Sun, 22 May 2016 23:46:39 +0000
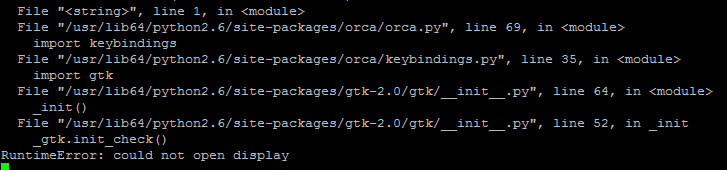
When I let Orca finish, I do get a .engrad file that is not empty, and orca terminates normally. I tried using your files, and I end up getting the same result as before, an empty .engrad file. When I try to run amber on the command line, I get the attached error. Could this be relevant at all?
________________________________________
From: Stanislav Simko <s.simko.uu.nl>
Sent: Sunday, May 22, 2016 4:31:58 AM
To: AMBER Mailing List
Subject: Re: [AMBER] QM/MM Interface
try to let finish the orca calculation - it shouldn't take very long
(depending on your QM region) to see if you obtain nonzero .engrad
file. This is critical part since Amber can not continue without this
file and it seems to me that there's smth wrong with orca calculation
since the file is created (i.e. orca most probably ran but crashed) but
it's empty. If that tells you nothing then try to run the attached
calculation (you need to prepare .prmtop file yourself though, it's too
big to attach) - i just tested it and it should run fine - the tpl file
is prepared from your .inp (i also attach you the submit script for SGE
batch).
best
stano
-- Stanislav Simko. PhD applicant Inorganic Chemistry and Catalysis
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Capture.PNG)
