Date: Mon, 2 May 2016 13:57:07 +0200
I did all atom simulation of my protein-ligand complex on 2 GPUs (Tesla
M2090). Here is my input file:
All atom simulation
&cntrl
imin = 0,
irest = 1,
ntx = 5,
ntb = 2,
ntp = 1,
cut = 8.0,
ntc = 2,
ntf = 2,
tempi = 298.0,
temp0 = 298.0,
ntt = 3,
taup = 2,
ig = 47123,
iwrap = 1,
gamma_ln = 1.0,
nstlim = 35000000, dt = 0.002
ntpr = 1000, ntwx = 1000, ntwr = 50000
&end
After 13399000 steps, simulation stopped and I got following error in
output file:
"Unit 16 Error on OPEN: prod3.rst"
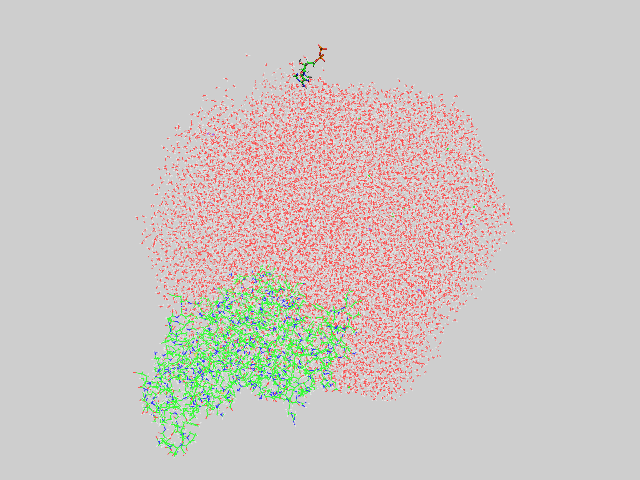
When I generated the pdb file using ambpdb, I observed that proteins are
ligands are outside the water-box (see attached image).
Can anyone suggest me what am I doing wrong.
*Thanks,Hirdesh*
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: error.png)
