Date: Fri, 11 Mar 2016 20:41:36 -0500
Dear all,
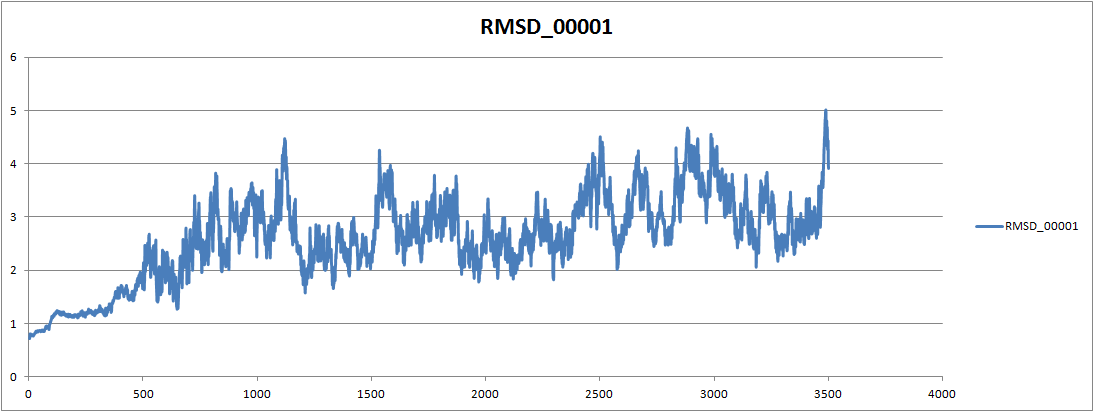
I just finished a 1 ns heating and a 60 ns production simulation. But when
I processed the RMSD I feel like it is too high. I know the fluctuation
from 1-3 angstron is expected but mine looks like it is over 4 sometimes.
Could you please explain what might have gone wrong during simulation? IS
there anything I can do to avoid such fluctuation during simulation? I
used exactly same input and time to run another simulation with a different
protein complex and rmsd was a lot lower in that case. I know it depends on
the system but if I could understand if there is anything I can do to
improve this it would be of great help. Can I even use any of these frames
to process mmpbsa script?
Rmsd input:
trajin heat.rmsd
trajin equil.rmsd
reference x.inpcrd
rms reference out backbone.rmsd .CA,C,N
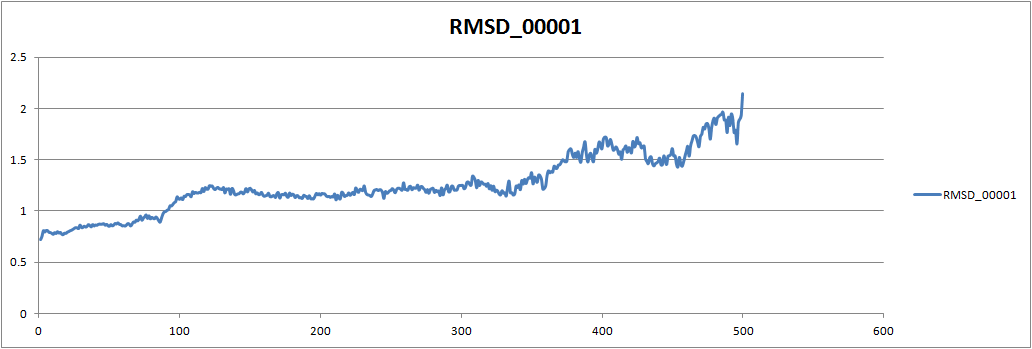
[image: Inline image 1]
If I remove equil.mdcrd an donly do trajin heat.mdrcd, rmsd is alot lower
but I can see it is increasing towards the end. So I think it is
fluctuating more during equilibrium stage. Following is the one only with
heat.mdcrd
[image: Inline image 2]
Thank you all in advance for your time!!!
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
(image/png attachment: 02-image.png)
