Date: Fri, 4 Mar 2016 10:10:03 +0530
Dear AMBER users,
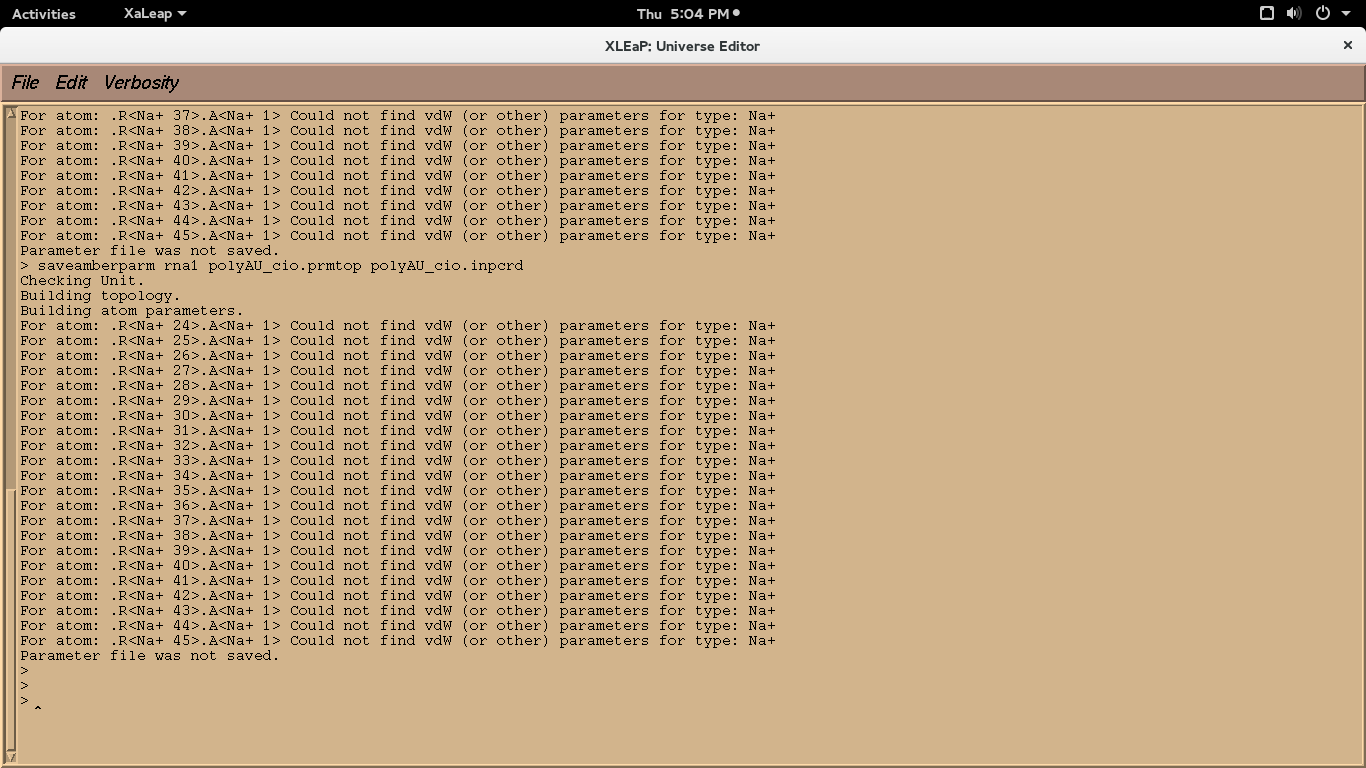
I'm new to amber. I have downloaded and installed AmberTools15 but i didn't
have licensed Amber 14. I have tried to run the tutorial " setting up
duplex DNA: polyA-polyT". I have used the "leaprc.ff14SB" force field and
"loadamberparams frcmod.ionsjc_tip3p" model. I could generate prmtop and
inpcrd files. But after adding ions while running "saveamberparm dna1
polyAT_cio.prmtop
polyAT_cio.inpcrd" for neutralised system, I'm getting following error.
Is it because of not having licensed Amber 14 or any other errors in input.
.
-- With kind regards, Sreemol
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot_from_2016-02-25_17-04-28.png)
