Date: Tue, 21 Jul 2015 12:55:17 +0800
Dear All,
I have run 2ns qm/mm on a stabilised ligand-protein complex using AMBER12.
I want to calculate the free binding energy. First I used the mm_pbsa.pl
using the script by Holger Gohlke. I modified the &GB section to include
the qm parameters. The output returned positive values for GBTOT which are
rather high.
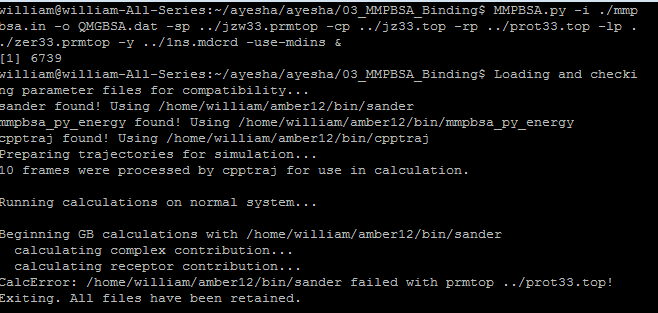
Then I tried to use the mmpbsa.py method and created the mmpbsa.in file as
indicated on the mailing list. The mdins files were created and as
mentioned on the mailing list to remove the "dec_verbose" and rerun the
mmpbsa. I did that however the error of CalcError: sander failed with
prmtop ../prot33.top! is coming.
Can someone guide me to set up?
Thank you
Ayesha Fatima
PhD candidate
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: error_msg.PNG)
- application/octet-stream attachment: _MMPBSA_gb_qmmm_com.mdin
- application/octet-stream attachment: _MMPBSA_gb_qmmm_lig.mdin
- application/octet-stream attachment: _MMPBSA_gb_qmmm_rec.mdin
- application/octet-stream attachment: mmpbsa.in
