Date: Thu, 16 Jul 2015 08:43:18 +0530
Hello Everyone
Firstly, I will briefly describe about what I am trying to do and then give
details about the error, so that it becomes easy to comprehend.
I am trying to perfomr an MD and the MM-PBSA calculations on a set of
Host-Guest complexes to estimate their binding strength.
Here the hosts are beta-cycledextrin and their suflobutylether and
2-hydroxypropyl derivative. And guests are various hydrophobic drug
molecules.
In the past I was successful in building the prmtop and inpcrd file for
beta-cyclodextrin and its 2-hydroxypropyl derivative using chimera.
But soon realized that prmtop and inpcrd generated using chimera had
missing information about the IFBOX, so i used to write the frcmod and the
lib files from parmed or Xparmed using the chimera generated prmtop and
inpcrd.
Later, I used to load the original pdb of the complex and the frcmod and
lib files in leap and once again add ions, neutralize and add water and
define box and save new prmtop and inpcrd and then could run MD and
subsequently MM-PB/GB-SA without any problem.
Now the problem is, I am unable to do the same for a set of new complexes
which involve suflobutylether and 2-hydroxypropyl derivative of
beta-cyclodextrin (as mentioned above).
Though i can save frcmod file, i am unable to save lib using WriteOFF
command in parmed.
The programs exits with the following error
> writeOFF case_2_new.lib
Writing Amber OFF file case_2_new.lib
Traceback (most recent call last):
File "/opt/amber14/bin/parmed.py", line 182, in <module>
parmed_commands.cmdloop()
File "/usr/lib64/python2.6/cmd.py", line 142, in cmdloop
stop = self.onecmd(line)
File "/usr/lib64/python2.6/cmd.py", line 219, in onecmd
return func(arg)
File "<string>", line 1, in <lambda>
File
"/opt/amber14/lib/python2.6/site-packages/ParmedTools/parmed_cmd.py", line
150, in _normaldo
action.execute()
File
"/opt/amber14/lib/python2.6/site-packages/ParmedTools/ParmedActions.py",
line 569, in execute
lib = ResidueTemplateContainer.from_structure(self.parm).to_library()
File
"/opt/amber14/lib/python2.6/site-packages/chemistry/modeller/residue.py",
line 233, in from_structure
rt = ResidueTemplate.from_residue(res)
File
"/opt/amber14/lib/python2.6/site-packages/chemistry/modeller/residue.py",
line 146, in from_residue
inst.add_atom(copy.copy(atom))
File
"/opt/amber14/lib/python2.6/site-packages/chemistry/modeller/residue.py",
line 92, in add_atom
raise ValueError('Residue already has atom named %s' % atom.name)
ValueError: Residue already has atom named O1
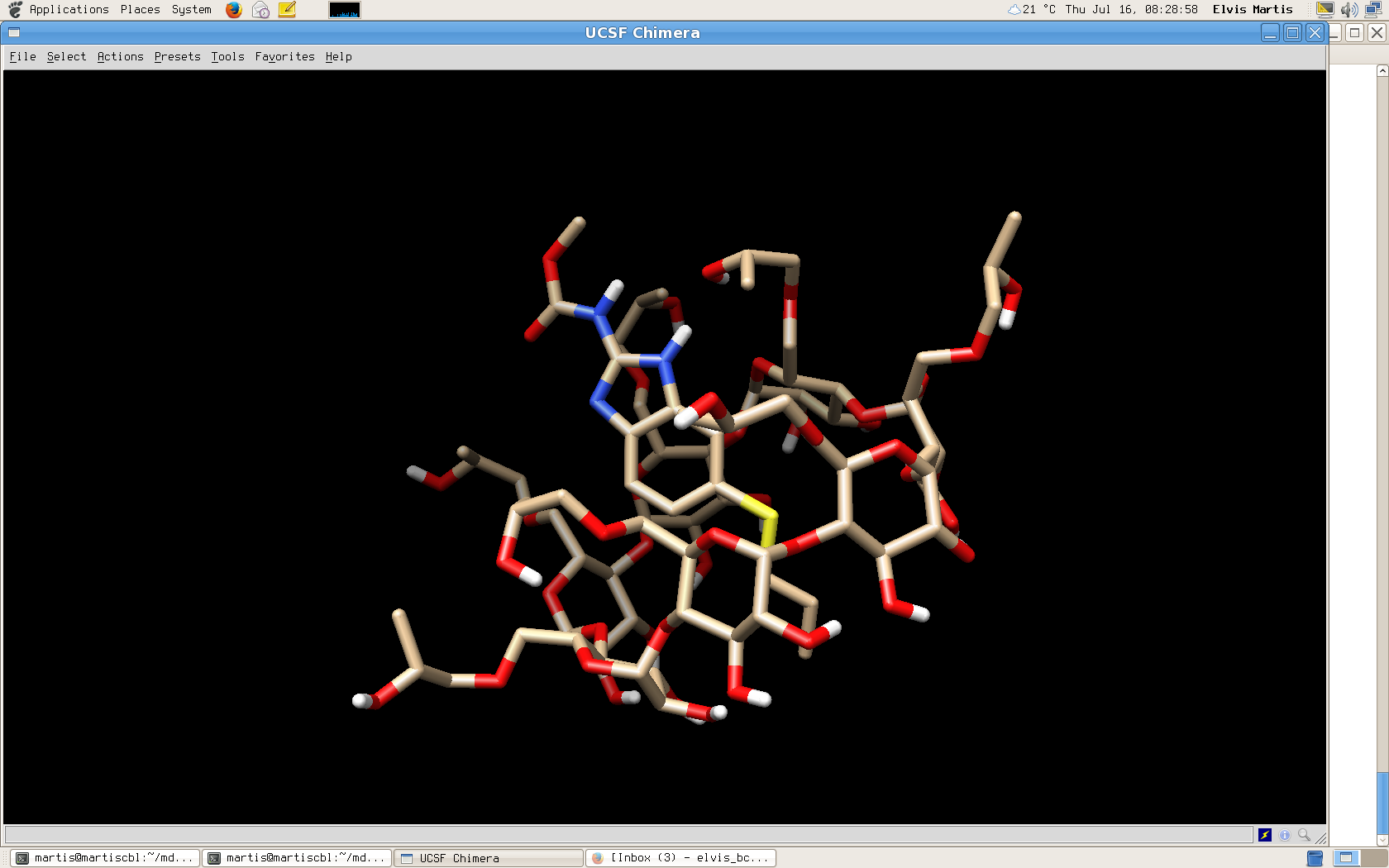
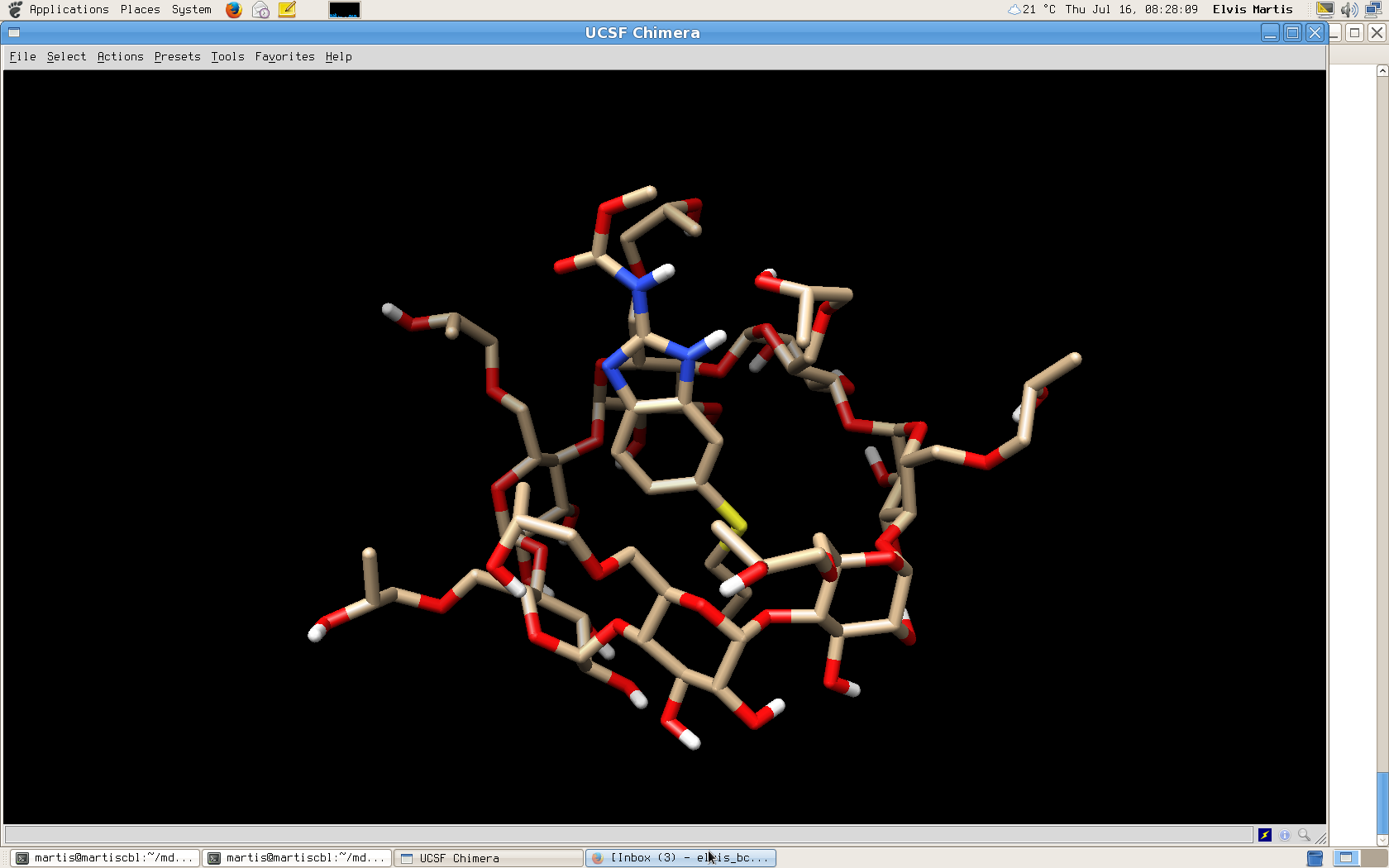
I am pasting the image of how my complex looks like.
[image: Inline images 1]
[image: Inline images 2]
Thanks in advance.
Regards
Elvis Martis
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
(image/png attachment: 02-image.png)
