Date: Wed, 15 Jul 2015 10:30:48 +0200
Hello:
I've got a protein/ligand complex system and I try to minimize them in
vacuum with following parameters:
&cntrl
imin = 1,
maxcyc = 5000,
ncyc = 250,
ntb = 0,
igb = 1,
cut = 12
/
FINAL RESULTS
NSTEP ENERGY RMS GMAX NAME NUMBER
4498 -1.1845E+04 2.5516E-02 4.1925E-01 C 2691
BOND = 214.7069 ANGLE = 836.2659 DIHED = 6010.7792
VDWAALS = -3445.4802 EEL = -26746.0685 EGB = -3225.0493
1-4 VDW = 1211.6324 1-4 EEL = 13298.6115 RESTRAINT =
0.0000
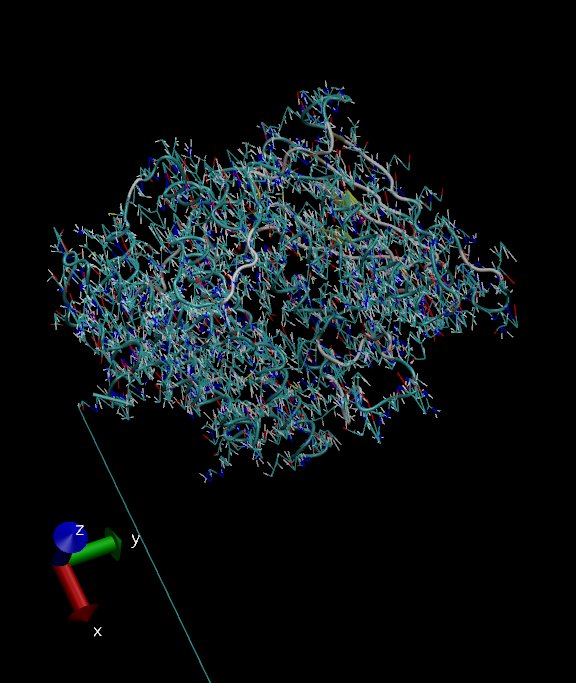
When I import the structure into VMD, I noticed that it looks very
strange: there are many atom clashes.... Here I've attached a screenshot
for it. The initial structure is a crystal structure and it was prepare
well before I set up the system.
Does anybody have any idea what's happening?
thx a lot
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: 1.jpg)
