Date: Tue, 26 May 2015 14:13:48 -0700
hello Amber community,
I am following the Amber Tutorial 16 on running lipid simulation and I am
in the equilibration stage with no restrains at all. Here is my input file:
&cntrl
imin=0,
ntx=5,
irest=1,
ntc=2,
ntf=2,
tol=0.0000001,
nstlim=2500000,
ntt=3,
gamma_ln=1.0,
temp0=303.0,
ntpr=5000,
ntwr=5000,
ntwx=5000,
dt=0.002,
ig=-1,
ntb=2,
ntp=2,
cut=10.0,
ioutfm=1,
ntxo=2,
/
/
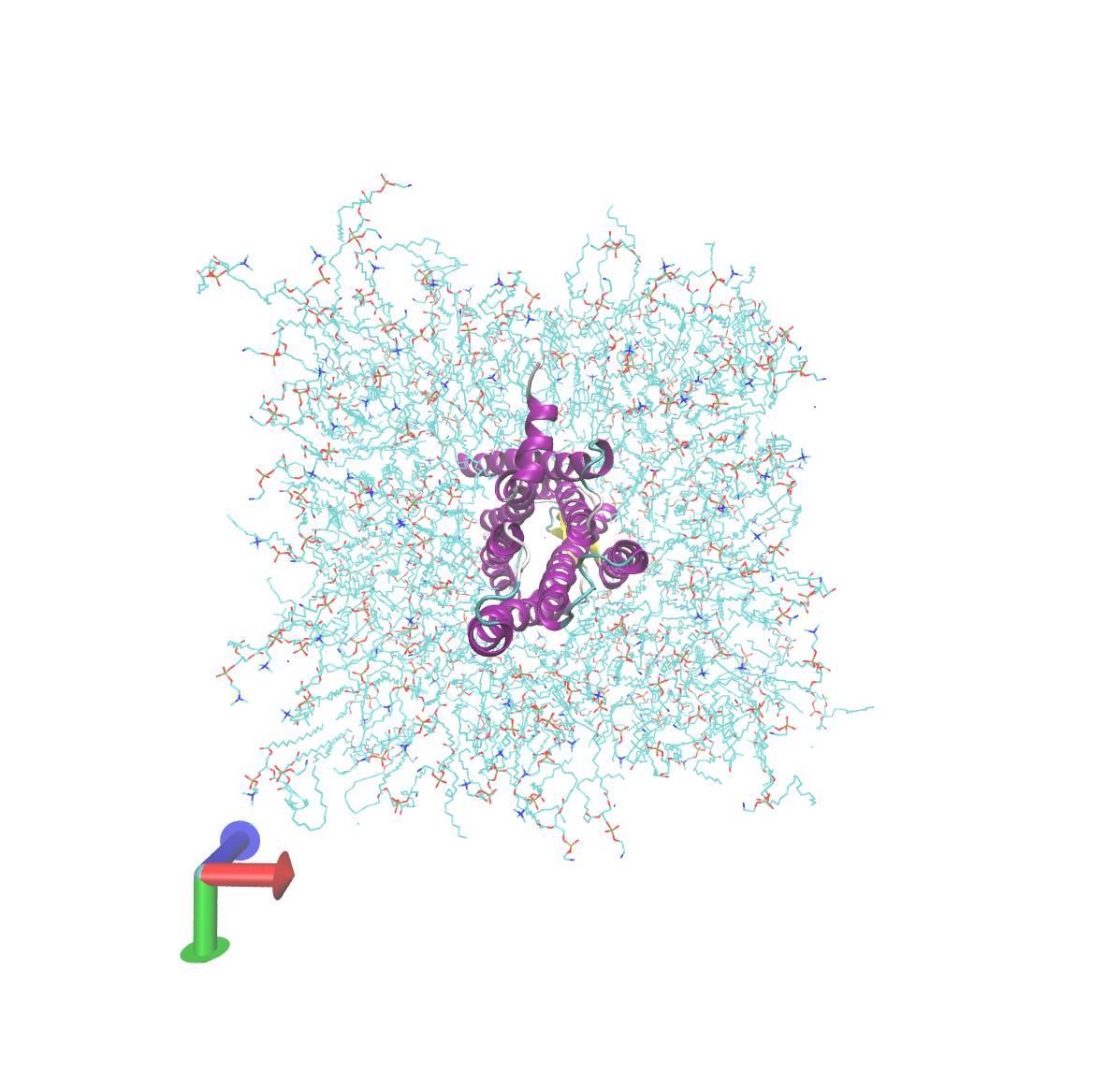
I am checking the output trajectory in VMD and I am not quite sure whether
the lipids look all right. They are kind of loose especially the ones on
the edge. When I visualize them dynamically, it feels like a few of them on
the boundary are "appearing" and "disappearing". I suppose that's the
periodic boundary box? The trajectory has been re-imaged using all the
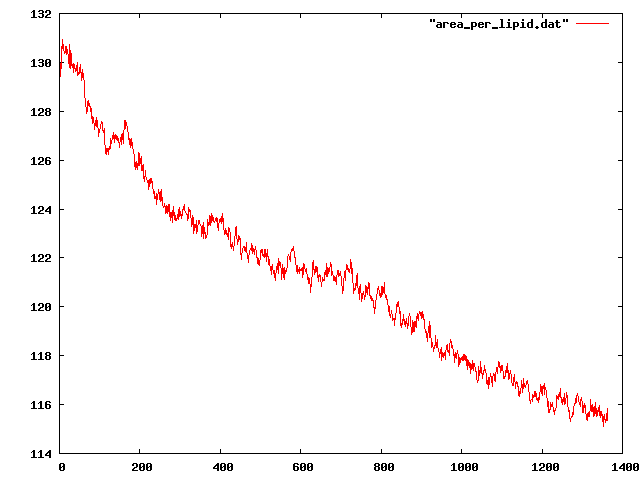
protein residues as centre. Also I calculated the "lipids per area" and
clearly they are not converged yet. But I am a bit surprised. Because in
the tutorial, lipids seem to have converged rather fast at the beginning of
the simulation.
I attached some pictures and look forward to any comments!
Thank you!!
Victor
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: printme.png)

(image/jpeg attachment: vmdscene.jpeg)
(image/jpeg attachment: vmdscene_1.jpeg)
