Date: Wed, 13 May 2015 10:51:48 -0700
hello all,
I am following tutorial 16 on lipid simulation. To confess, I did not
follow the exact same procedure. For the "hold" step, I only ran it once
instead of the 10 times in the tutorial. My argument is that I am doing
cpu-based simulation.
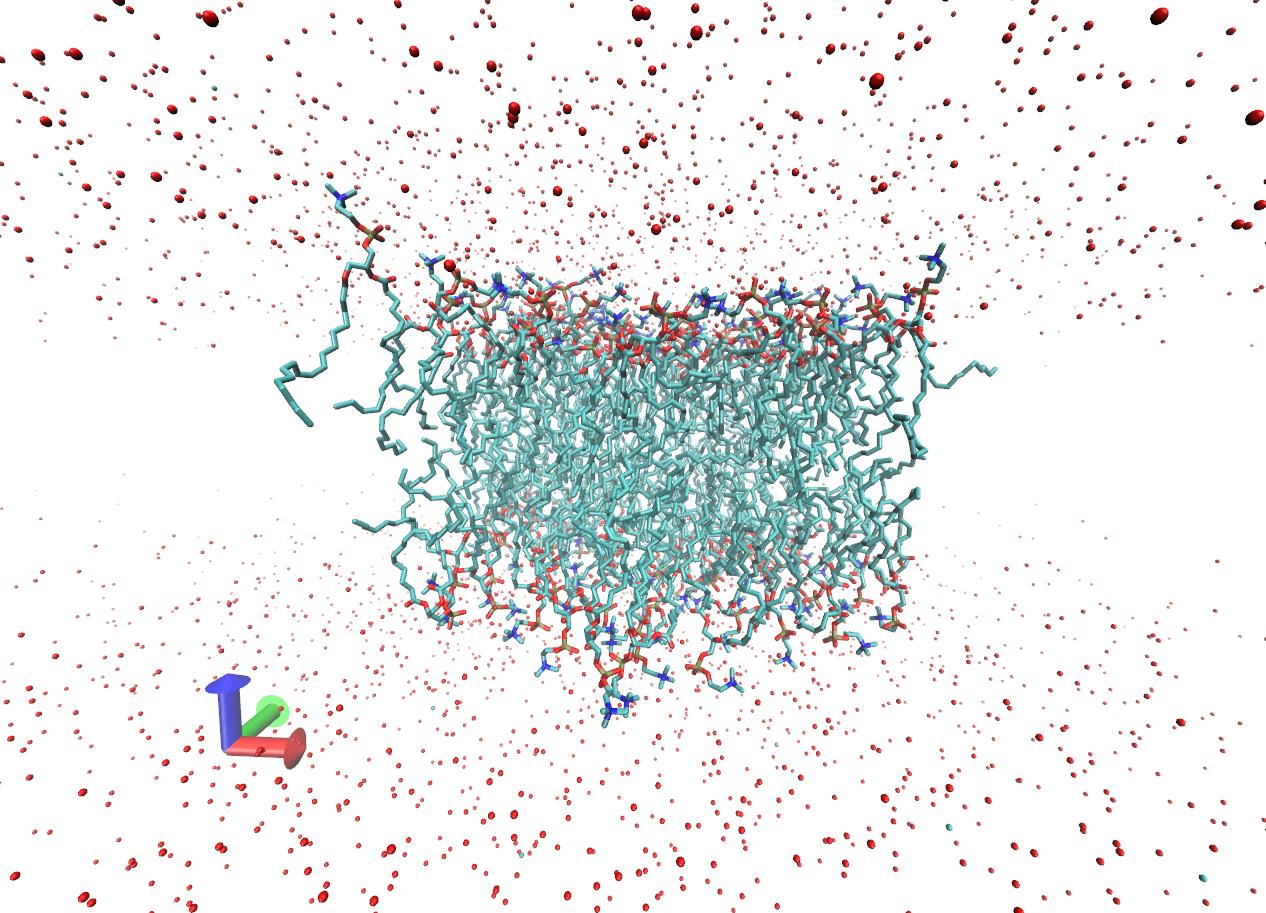
Now I am checking the results and I see that in my system, the water
molecules are naturally "spreading out" like in a soluble protein system.
Please see the picture in attachment. However the picture in tutorial
(after 5ns of production run) looks different. In that picture, all the
water molecules seem to be still stacking on top and bottom of the lipids
just like when I first set up the system without running it. I am a little
confused here. Can someone please clarify this for me? Thank you.
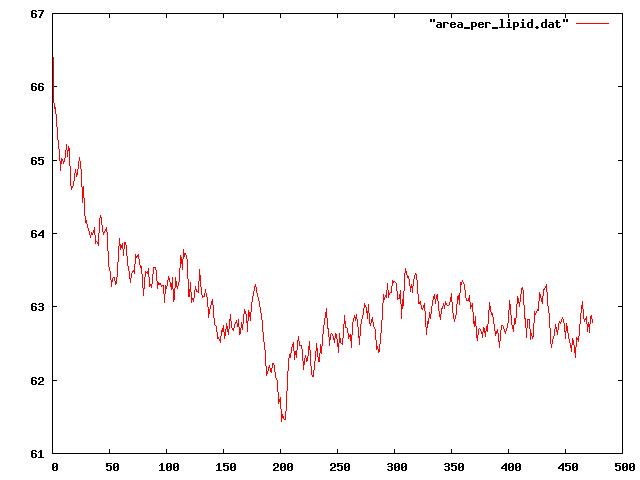
To provide more information, I also attached the plot of area per lipid.
Thank you !!!
Victor
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: lipids.jpeg)
(image/png attachment: printme.png)
