Date: Tue, 3 Mar 2015 18:05:57 -0500
Hello all,
I am a new Amber user and so I followed and completed Tutorial B0 up to the
point of VMD visualization of the result.
I am now trying to repeat the tutorial up to this point with a prmtop and
inpcrd files that I produce using tleap and a customized frcmod file. Here
are the steps I take:
1) Read the .mol2 file that I have produced outside amber and produce a
corresponding frcmod file for it
The following are done in tleap:
2) source leaprc.ff12SB
3) mol = loadMol2 foo.MOL2
4) loadAmberParams frcmod.foo
4.5) check mol (no errors are detected)
5) solvateBox mol TIP3PBOX 10.0
6) saveAmberParm mol prmtop inpcrd
Then I try to run through tutorial B0 using these newly produced prmtop and
inpcrd and the exact same 01_Min.in, 02_Heat.in, 03_Prod.in files and
commands given in Tutorial B0:(
http://ambermd.org/tutorials/basic/tutorial0/#Getting_started_with_Linux_).
However this fails after the Run Minimization command and gives the
following error: Segmentation fault (core dumped). First I tried this with
a .MOL2 file for acetic acid, guanidinium etc. Eventually I created a .MOL2
file for the same molecule used in the tutorial and produced the prmtop and
inpcrd files using the same steps 1-6 as above.
This time with I was able to complete the tutorial. I then tried to modify
the molecule by removing the methyl on the central carbon and completed
steps 1-6 and ran through the tutorial again with no trouble. I then
removed the atoms on one side of the molecule, completed steps 1-6 and ran
through the tutorial again with no problem. By now the structure was
extremely close to acetic acid so I modified it again to be acetic acid but
this time the error again appeared at the Run minimization step of the
tutorial.
What is causing the tutorial to fail when I use a prmtop and inpcrd file
produced from a .MOL2 file for a molecule of certain size but causing it to
fail for others? And how would I fix it? The problem is particularly
puzzling since neither the *.in files or commands in Tutorial B0 seem to
make any reference to molecule size or shape.
I have attached images of the molecules with which I was able to complete
the tutorial using prmtop and inpcrd files produced with steps 1-6.
Thank-you for your help.
Sanmeet
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
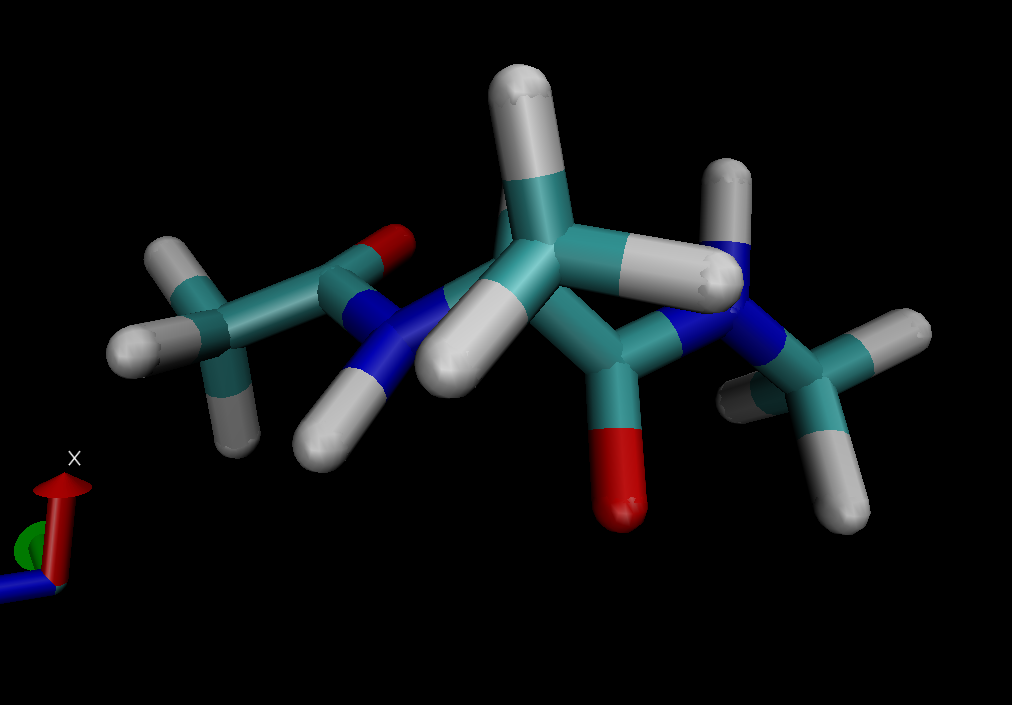
(image/png attachment: original-molecule.png)
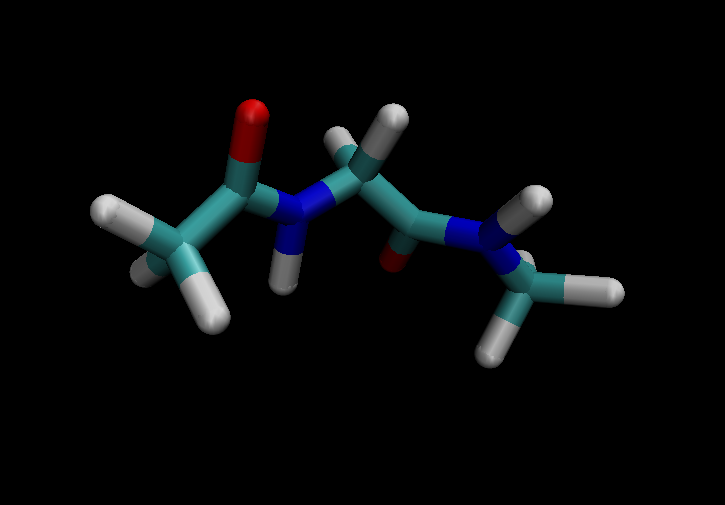
(image/png attachment: methyl-on-central-carb-removed.png)
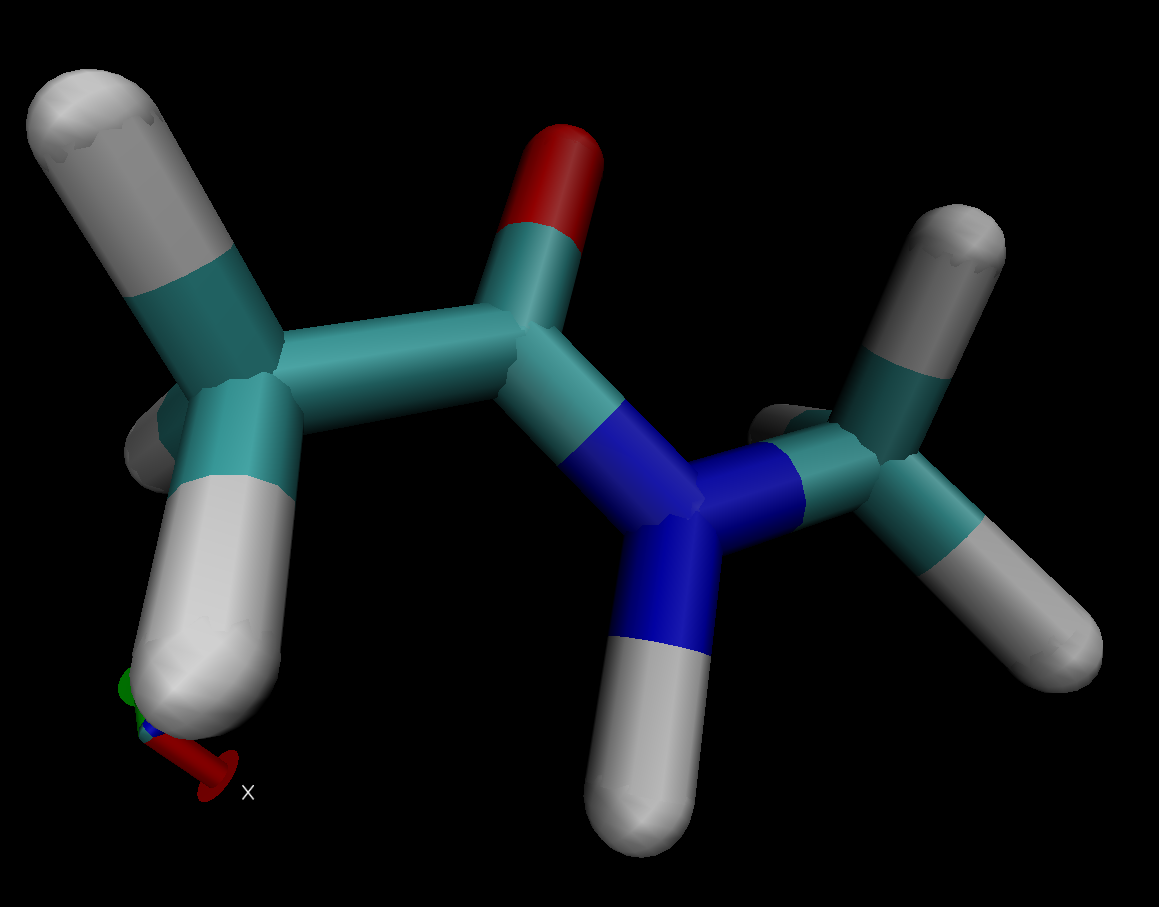
(image/png attachment: atoms-on-one-side-removed.png)
