Date: Thu, 8 Jan 2015 03:06:36 +0200
Dear sir
I wanted to run MD simulation for a protein in a TIP3P water box with
average 7100 atoms for 30 ns with step length of 0.001 ps. I am using NPT
ensemble with Langevin dynamics and periodic boundary conditions.
I have the following control file !
imin=0, nmropt=0,
ntx=5, irest=1, ntrx=1,
ntxo=1, ntpr=100, ntwr=100,
iwrap=1, ntwx=100, ntwv=0, ntwe=0,
ioutfm=0, ntwprt=0, idecomp=0,
ntf=2, ntb=2, igb=0, nsnb=1,
ipol=0, gbsa=0, iesp=0,
dielc=1.0, cut=10.0, intdiel=1.0,
ibelly=0, ntr=0,
nstlim=5000, nscm=500, nrespa=1,
t=0.0, dt=0.001, vlimit=20.0,
ntc=2, jfastw=0, tol=0.00001,
Tempi=310.0 , Temp0=310.0 , ntt=3, tautp=0.5, gamma_ln=2,
pres0=1.0, ntp=1, taup=2,
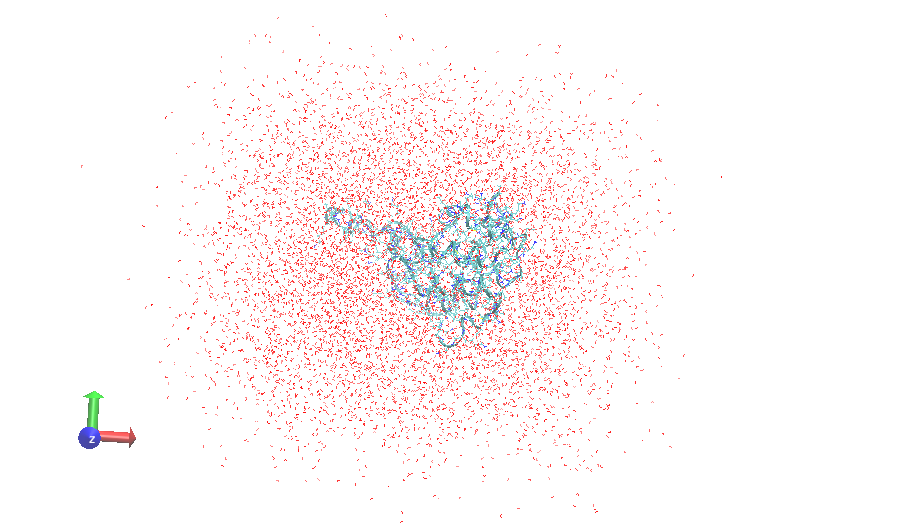
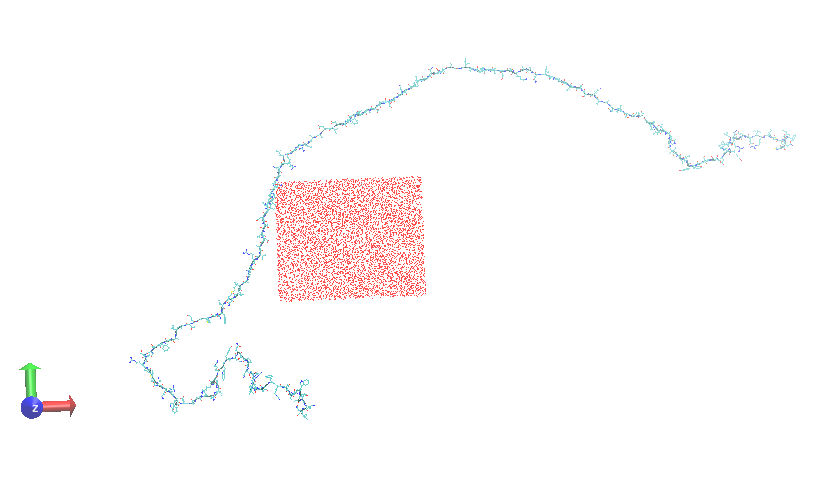
However through the data analysis of the 30, I found that the protein
underwent complete unfolding leaving the water box although the boundary
conditions. Kindly find the attached snapshofts of the protein before and
after complete unfolding. what do you think the source of error in my
simulation or control file ? I can support all the information that might
help figuring out the sources of error . I will appreciate your prompt
response.
Thanks in Advance !
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 2f47_0000md.png)
(image/png attachment: 2f47_6523md.png)
