Date: Wed, 10 Sep 2014 20:09:22 +0800 (CST)
Hi all,
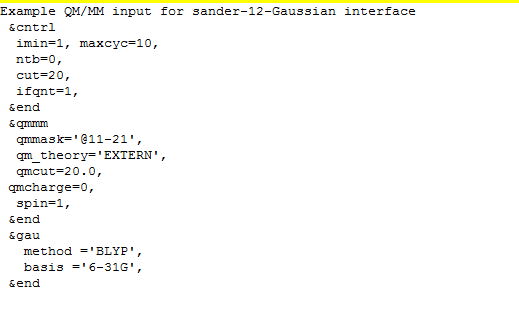
Recently, I want to optimize the molecule. I use the Amber to simulate the atom in MM region, and use the Gaussian to simulate the atom in QM region. But there is no MM atoms in the Log file of outfile , It just has the atom in QM region.
Thank you in advance.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: ______1.png)
- text/plain attachment: old.gau_job.log
