Date: Thu, 14 Aug 2014 17:23:13 +0800 (GMT+08:00)
Dear all,
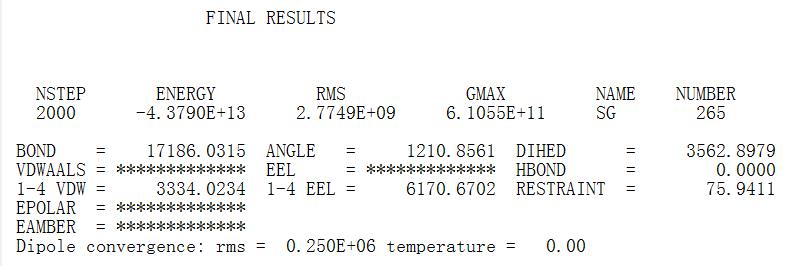
Recently, I've been working on a job about using a polarized FF(leaprc.ff02) to simulate a protein-nanotube system(model processing is as A00). When I run minimization of step one, mdout results seemed to be fine(A01). When I went to step two, however, I found that the RMS and dipole convergence rms became very large(The mdout file is as below A02. ). With many uncertainties I went on with the minimization of step three, and I shockingly found that the RMS and dipole convergence rms turns out to be too large to see(The mdout file is as below A03)!! I have tried many different combinations of namelists in the $cntrl, leading to the same results. When I turned to the amber tutorials hoping to find some helpful answers but I got nothing related to this. So, could you help me with this? All in all, I hope you can give me some suggestions about using amber polarized FF.
Regards,
Terry_Zhang
A00:
source leaprc.gaff
source leaprc.ff02
loadamberparams IC6.frcmod
comp=loadpdb all.pdb
solvatebox comp TIP3BOX 10.0
addions comp Cl- 0
set default ipol=1
saveamberparmpol comp in.prmtop in.inpcrd
A01:
Minimize
&cntrl
imin=1,maxcyc=2000,ntmin=2,
cut=10.0,
ntb=1,
ipol=1,
ntr=1,
restraint_wt=500,
restraintmask=':1-106'
/
A02:
Minimize
&cntrl
imin=1,maxcyc=2000,ntmin=2,
cut=10.0,
ntb=1,
ipol=1,
ntr=1,
restraint_wt=500,
restraintmask=':1-104.CA,N,C,O | : 105-106'
/
A03:
Minimize
&cntrl
imin=1,maxcyc=2000,ntmin=2,
cut=10.0,
ntb=1,
ipol=1,
ntr=0,
/
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber

(image/jpeg attachment: CMC32D8.tmp.JPG)
(image/jpeg attachment: CMC9B8B.tmp.JPG)

(image/jpeg attachment: CMC8F4E.tmp.JPG)
