Date: Thu, 6 Feb 2014 01:14:42 +0800
Respected sir,
Thanks for the reply. This has certainly made few things clearer than before.
After I run following command
antechamber -i ligand.pdb -fi pdb -o ligand.mol2 -fo mol2 -c bcc -s 2 -nc 0
I get sqm.pdb i.e. pdb file of the ligand generated after SQM, in addition to ligand.mol2.
This was followed by
parmchk -i ligand.mol2 -f mol2 -o ligand.frcmod
the following section of leaprc was used to merge ligand with protein
UNK = loadmol2 ligand.mol2
loadamberparams ligand.frcmod
saveoff UNK ligand.lib
saveamberparm UNK ligand.prmtop ligand.inpcrd
receptor = loadpdb protein.pdb
saveamberparm receptor receptor.prmtop receptor.inpcrd
savepdb receptor receptor.pdb
saveoff receptor receptor.lib
comp = combine {receptor UNK}
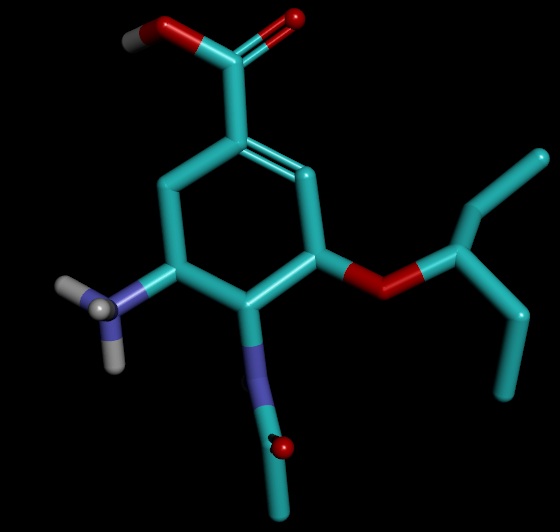
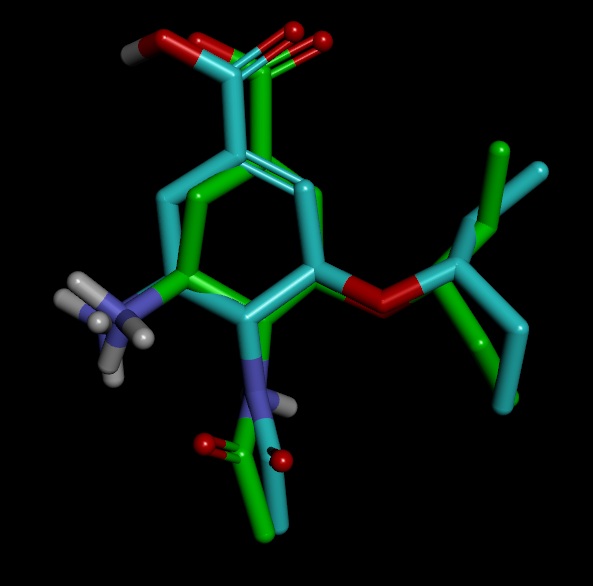
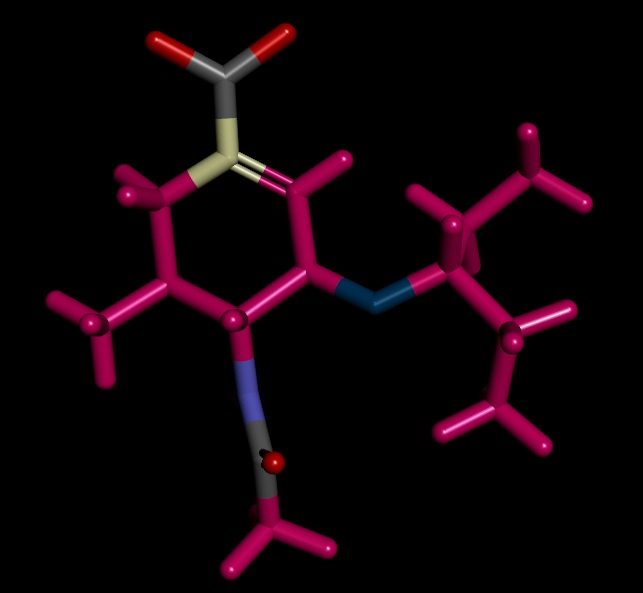
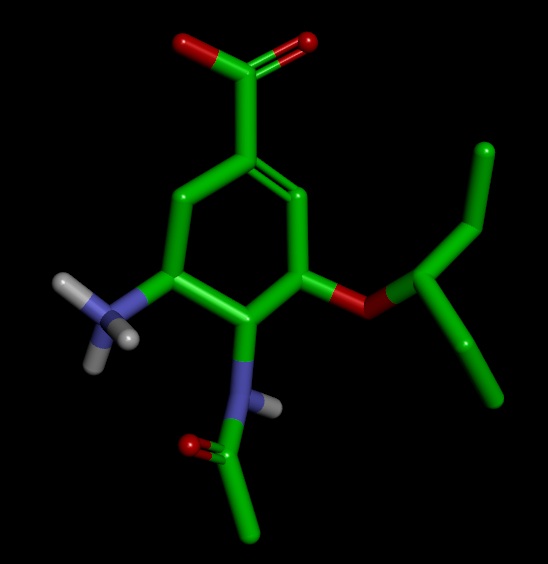
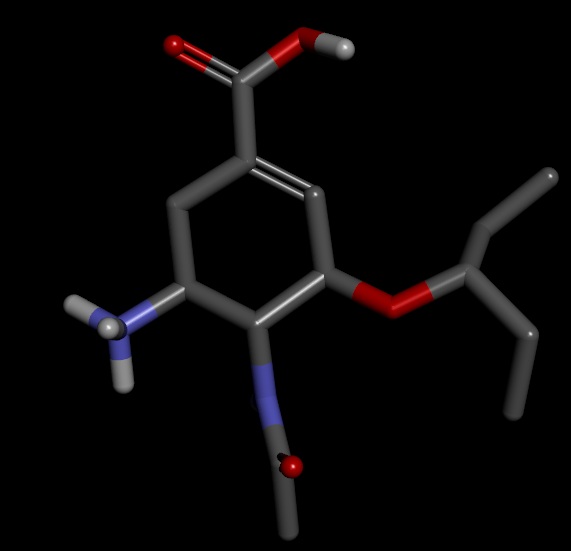
On close examination I got confused in few things. I have attached the images of the structures for better understanding
1) the ligand.mol2 produced by antechamber has OCO group instead of OC=O. However, the final complex has required group OC=O group though ligand.mol2 file was read to merge with receptor.
2) The ligand.mol2 overlapped with original ligand.pdb file perfectly.
3) As expected the structure of sqm.pdb is different from original.pdb
4) if ligand.mol2 is close to original structure instead of sqm.pdb then what is the use of sqm.pdb file. Am I right in assuming that geometry optimization has not been achieved in real sense.
5) will it be a good idea to use sqm.pdb instead of ligand.mol2 to combine with receptor in order to form complex? i.e. UNK = loadpdb sqm.pdb instead of UNK = loadmol2 ligand.mol2
Also it will be really helpful if you can pass me some references related to partial atomic charge determination and ligand/residue model building.
Thanks and regards,
Nitin Sharma
-----Original Message-----
From: Karl Kirschner [mailto:k.n.kirschner.gmail.com]
Sent: Wednesday, January 29, 2014 5:14 PM
To: AMBER Mailing List
Subject: Re: [AMBER] optimise ligand geomatry before MD
Hello Nitin,
Yes, in general one should optimize a ligand's geometry prior to an MD simulation. If the force-field parameters are present and trustworthy, then performing an MM minimization is sufficient. This is important so that you do not introduce artificially large forces, arising from the molecule residing at a high energy state on the MM's potential energy surface, at the start of your simulation.
However, if the parameters are not present (e.g. torsion terms, partial atomic charges) then you will need to optimize the ligand using QM or Antechamber (i.e. sqm). Most Amber force fields were developed with geometries optimized at HF/6-31G(d), but many use the Antechamber/sqm method for optimizing ligands, which I believe is semiempirical. Both methods are comparable, in the sense that they optimize the geometry, but differ in the theory used to do so. Different theories will lead to different optimized geometries, sometimes they have relatively minor differences (e.g. bond distances) and sometimes they are more significant (e.g. different rotamers).
Once you have the optimized conformation, then you use it as an input to determine partial atomic charges. There is quite a bit of literature for this already present for Amber, including using multiple conformations to determine a single charge set for a molecule.
It is reasonable/necessary to optimize a ligand's conformation that you obtained from a docking pose. If you used flexible docking methods, then note that this conformation may change due to the absence of the binding pocket.
As for websites/methods that you can use, Antechamber's can perform an optimization (via sqm) and to determine partial atomic charges. The R.E.D.
website will also allow you to do both in a manner that is balanced with the Amber force fields. You can also have the option of downloading a number of QM software packages, such as Gamess and Gaussian. I would suggest doing some more reading regarding partial atomic charge determination and ligand/residue model building (e.g. Parm94, Gaff, Glycam06, R.E.D., Wolf2Pack, etc. papers).
Best regards,
Karl
On Tue, Jan 28, 2014 at 7:44 PM, Nitin Sharma <sharmanitin.nus.edu.sg>wrote:
> Dear amber users,
>
> Is it necessary to optimize ligand geometry before molecular dynamics
> simulation?
>
> I am using AMBER for MD simulation.
>
> I have seen studies where ligand was parameterized according to
> quantum chemical calculations, which included performing a geometry
> optimization with Gaussian98 at the Hartree-Fock/6-31G* level before
> determining atomic charges and atomic types using Antechamber module
> of the Amber molecular dynamics software package.
>
> However, i have seen studies where ligand were assigned generalized
> Amber force field (GAFF) atom types and AM1-BCC atomic charges
> obtained by adding the bond charge correction (BCC) using Antechamber.
>
> Are both protocols comparable ?
>
> As i am thinking to use pose generated as result of docking will the
> ligand geometry optimization be good idea?
>
> can someone also tell me web-server or software to do ligand geometry
> optimization for AMBER MD simulation
>
>
> Thanks and regards,
> Nitin Sharma
> Department of Pharmacy,Faculty of Science,National University of
> Singapore,block S7, Level 2, Singapore : sharmanitin.nus.edu.sg<mailto:
> sharmanitin.nus.edu.sg> ; http://www.linkedin.com/in/imsharmanitin
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: complex.jpg)
(image/jpeg attachment: complex_SQM_overlap.jpg)
(image/jpeg attachment: ligand_mol2.jpg)
(image/jpeg attachment: ligand_sqm.jpg)
(image/jpeg attachment: original.jpg)
