Date: Mon, 6 Jan 2014 18:05:05 +0000
Hi Jason (and others on the AMBER list),
I check the gaff.dat file first to make sure that there was no dihedral term that contained "-cc-ca-" or "-ca-cc-". The reason I think the first term isn't being applied is that I used a scan script by S. Ruiz that I think is similar to the scan.sh script that you posted to this forum earlier. When I use the script, the relative energy plot that comes out looks just like the vdW/electrostat curve (from setting dihedral PK/idivf to zero) plus the second term [3.5/4*(1+cos(2x-180))]. When I plot the relative energy from the scan using my modified .frcmod file in excel and then manually add the second term [1.4/4*(1+cos(x))], it matches the QM result very well.
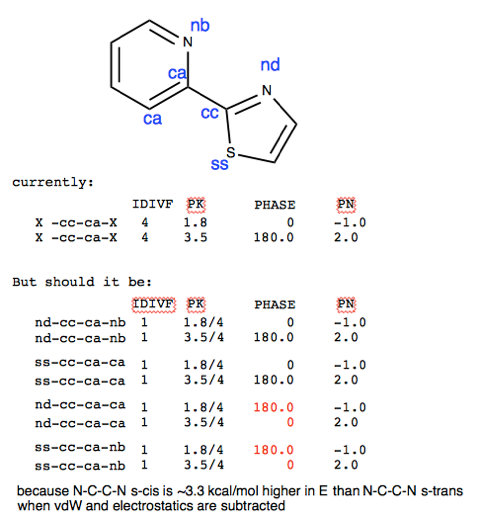
I have another question about setting up the dihedrals that may or may not be pertinent to my previous query. I'm looking at a pyridine-thiazole dihedral where the N-C-C-N' is ~3.3 kcal/mol higher in E in the s-cis conformation compared to the s-trans conformation, even when the electrostatic and vdw contributions are subtracted. Should I change my dihedral parameters accordingly (see attached .png file)?
Thanks,
Scott
[cid:f8c9c1d4-6de0-4b0f-bc19-a360ff283c9c.ad.unc.edu]
On Jan 6, 2014, at 12:32 PM, Jason Swails wrote:
>
> On Mon, 2014-01-06 at 00:52 +0000, Allen, Scott Edward wrote:
>> Hello,
>>
>> I'm trying to parametrize a dihedral in my small molecule. I find that when I incorporate my results into my .frcmod file, AMBER (tleap?) only reads the second term of the X -ca-cc-X dihedral (the phase=180 term). Has anyone had a similar problem? The full text of the .frcmod file is given below.
>
> What makes you say this? Did you use ParmEd or rdparm to verify that
> the first term is missing from the relevant torsion terms?
>
> Also note that "X -ca-cc-X " is only used when a specific torsion is not
> present in any of the parameter databases. It's possible that one of
> the terms you hoped to modify with your torsion wild-card has a specific
> definition in the gaff.dat file. In this case, the only way to override
> that torsion term is to use specific torsions (i.e., with full atom
> types as the first and last atoms rather than wildcards).
>
> HTH,
> Jason
>
>>
>> Thank you,
>> Scott Allen
>>
>> remark goes here
>> MASS
>>
>> BOND
>>
>> ANGLE
>>
>> DIHE
>> X -ca-cc-X 4 1.800 0.000 -1.000 from GA
>> X -ca-cc-X 4 3.500 180.000 2.000 from GA
>> cc-ss-cc-cd 1 1.100 180.000 2.000 same as X -c2-ss-X
>> cc-ss-cc-h4 1 1.100 180.000 2.000 same as X -c2-ss-X
>> cc-ss-cc-nd 1 1.100 180.000 2.000 same as X -c2-ss-X
>> cc-ss-cc-ca 1 1.100 180.000 2.000 same as X -c2-ss-X
>>
>> IMPROPER
>> ca-nd-cc-ss 1.1 180.0 2.0 Using default value
>> cc-h4-cd-nd 1.1 180.0 2.0 Using default value
>> cd-h4-cc-ss 1.1 180.0 2.0 Using default value
>> ca-cc-ca-nb 1.1 180.0 2.0 Using default value
>> ca-ca-ca-ha 1.1 180.0 2.0 General improper torsional angle (2 general atom types)
>> ca-h4-ca-nb 1.1 180.0 2.0 Using default value
>>
>> NONBON
>>
>
> --
> Jason M. Swails
> BioMaPS,
> Rutgers University
> Postdoctoral Researcher
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: dihedrals_problem.png)
