Date: Thu, 3 Oct 2013 12:57:43 +0000
Hi,
I encountered a problem in the VDW process when I used thermodynamic integration method to
calculate relative free energies similar to amber tutorial A9.
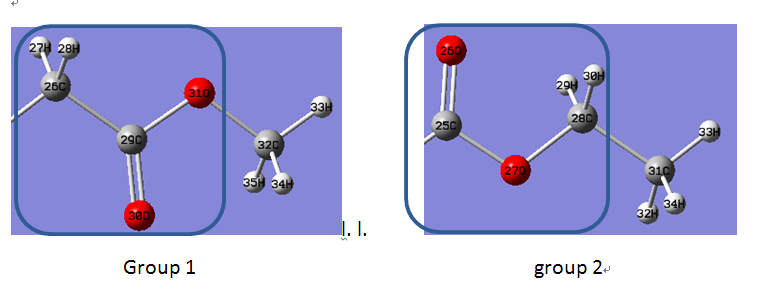
In fact, I put the following two groups into a cage, and calculate the relative binding free energies.
The two groups (attachment) have same atoms, and are just different from the position of carboxyl.
I calculate also through three steps: 1)remove change of group 1; 2)change group 1 to group 2;3)appear charge of group 2.
My prm and rst files seems all right, and I’m sure that the common atoms have same coordinates and same order in rst files.
There are six unique atoms in VDW process. My input files have defined the scmask and crgmask as:
V0
crgmask=”:TWO.C9,H18,H19,C10,O1,O2”
scmask=”:TWO.C9,H18,H19,C10,O1,O2”
V1
crgmask=”:REE.C9, O1,O2,C10,H18,H19”
scmask=”:REE.C9, O1,O2,C10,H18,H19”
Then I run sander, to minimization, equilibrium, and MD production. The minimization finished normally (but …). When sander came to equilibrium, the error appeared as:
Softcore Mask :TWO.C9,H18,H19,C10,O1,O2; matches 6 atoms
this run corresponds to V0, its softcore atoms interact fully for lambda=0
this process: 9257 atoms, partner process: 9257 atoms
Checking for mismatched coordinates.
WARNING: Local coordinate 1246 differs from partner coordinate 1246 !
Deviation is small, changing partner coordinate.
WARNING: Local coordinate 1247 differs from partner coordinate 1247 !
Deviation is small, changing partner coordinate.
WARNING: Local coordinate 1248 differs from partner coordinate 1248 !
Deviation is small, changing partner coordinate.
WARNING: Local coordinate 1249 differs from partner coordinate 1249 !
SANDER BOMB in subroutine sc_check_and_adjust
Atom coordinate disagreement
Also, I saw the rst file of minimization. I found that coordinates of common atoms became different between V0 and V1, even though they have same coordinates before minimization.
That is what the WANING says, that 1246-1249 are coordinates of common atoms in V0 and V1, they are same before minimization but different after minimization. In my experience, coordinates of common atoms could be change but keep the same values in V0 and V1 in these TI processes.
I guess the error come from the scmask string. How the unique atoms order in V0 and V1 files since they have completely different positions.
I don’t know if I express my problem clearly, you can ask any details about the calculation. I want to know the solution very much. I beg your answers.
Best regards,
Dandan Yuan
NanJing University
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 0and1.png)
