Date: Tue, 1 Oct 2013 21:11:22 +0800
Thanks for the suggestions Jason. But I reconfirmed that I have not
reversed the X-axes. When I plot the dihedral angles sampled from the 2
simulations (plots attached), I also see less sampling around the dihedral
angles which correspond to the barriers in those PMF plots.
Regards,
mohan
On Tue, Oct 1, 2013 at 7:50 PM, Jason Swails <jason.swails.gmail.com> wrote:
> On Tue, Oct 1, 2013 at 7:11 AM, Mohan Pradhan <pradhanmohan56.gmail.com
> >wrote:
>
> > Dear amber users,
> >
> > I am following the amber tutorial-
> > http://ambermd.org/tutorials/advanced/tutorial17/section1.htm for the
> > umbrella sampling of a dihedral angle of an alpha helical peptide from
> cis
> > to trans conformation.
> >
> > The commands and the WHAM meta file are exactly same as given in this
> > tutorial. The only change is that, in the 1st simulation, dihedral angle
> of
> > starting structure is 0 degree (from crystal structure) and then we
> sampled
> > 3 degree windows from 0 to 180 serially; while in 2nd simulation,
> dihedral
> > angle of starting structure is the 180 degree (structure extracted from
> 1st
> > simulation) and then we sampled 3 degree windows from 180 to 0 serially.
> > Each window is sampled for 500 ps in both the simulations. The PMF
> derived
> > from these calculations is quite contradictory to each other (plots
> > attached). Also, I ensure sufficient overlap of sampled dihedral angles
> > between consecutive windows.
> >
> > Any suggestions why the PMF calculated from these 2 simulations is not
> > similar (even qualitatively)?
> >
>
> They look qualitatively very similar. In fact, one looks like the mirror
> image of the other (reflected in the Y-axis). This suggests to me that
> what you are calling 180 degrees in the first plot you are calling 0
> degrees in the second (and vice-versa). Assuming you got one of the X-axes
> reversed, the profiles are not only qualitatively similar but probably also
> agree quantitatively within the uncertainty of each window (MBAR can help
> you compute uncertainties, as can bootstrapping with WHAM) -- especially
> with only 500 ps of simulation per window.
>
> Note that umbrella sampling does not have a 'directional' property to it
> like other methods like steered MD. Each window is sampling from an
> equilibrium distribution defined by a potential biased with an umbrella
> potential along a reaction coordinate. As a result, all snapshots that you
> use in the analysis should be from that equilibrium distribution (which
> means you should equilibrate each window and discard these 'equilibration'
> snapshots).
>
> HTH,
> Jason
>
> --
> Jason M. Swails
> BioMaPS,
> Rutgers University
> Postdoctoral Researcher
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
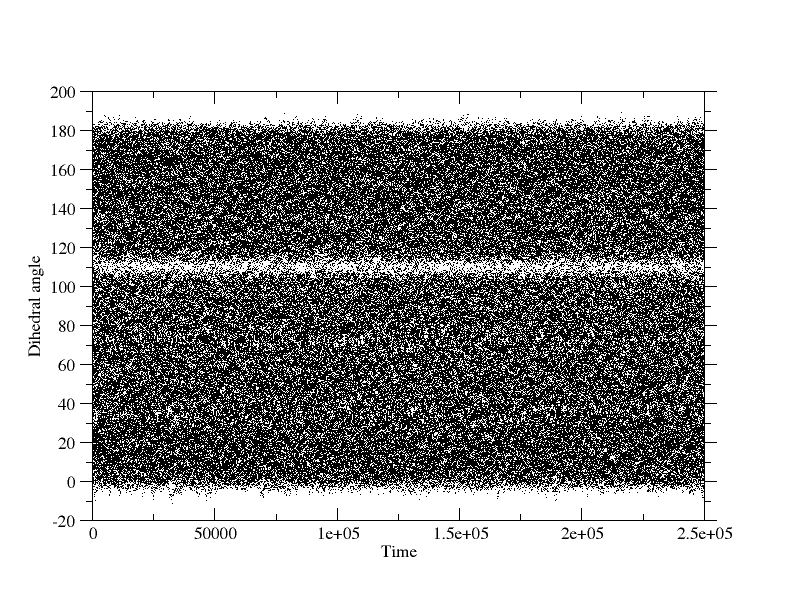
(image/png attachment: 0-180_all_dihedrals.png)
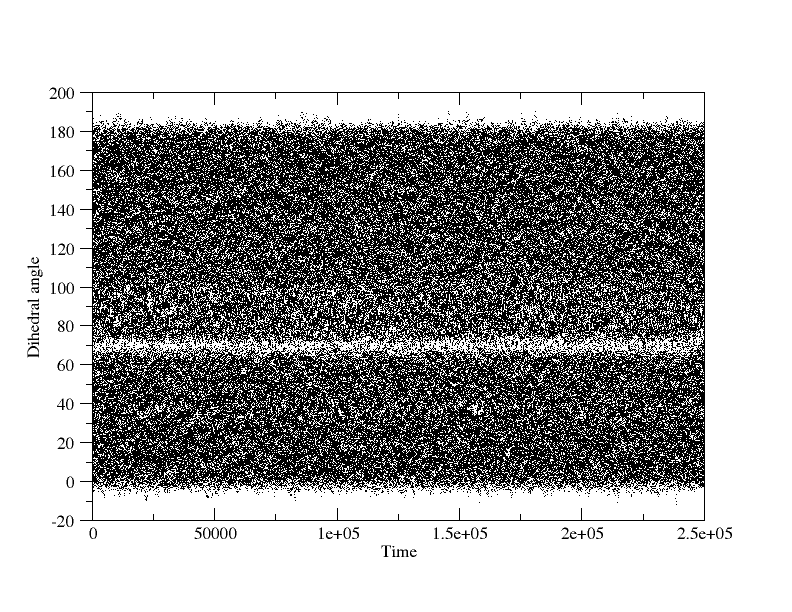
(image/png attachment: 180-0_all_dihedrals.png)
