On Fri, Sep 20, 2013 at 1:25 AM, Sumra Wajid Abbasi 30-FBAS/MSBI/F09 <
sumra.msbi30.iiu.edu.pk> wrote:
> Dear Smith
>
> Yeah you are right i loaded the ligand into VMD using Coordinates With
> Periodic Box info. now i load without Periodic Box info and image looks
> fine..But still calculation issue is there..
>
Upgrade to AmberTools 13 and see if the issue persists. If the problem is
fixed under the latest (free) version, it is unlikely to be fixed in older
(unsupported) versions.
HTH,
Jason
>
> Regards
>
> Sumra Wajid Abbasi
> PhD Scholar
> Computational Biology Lab, National Center for Bioinformatics
> Quaid-e-Azam University, Islamabad-45320 Pakistan.
>
>
> On Fri, Sep 20, 2013 at 6:05 AM, <wmsmith.uci.edu> wrote:
>
> > Hello,
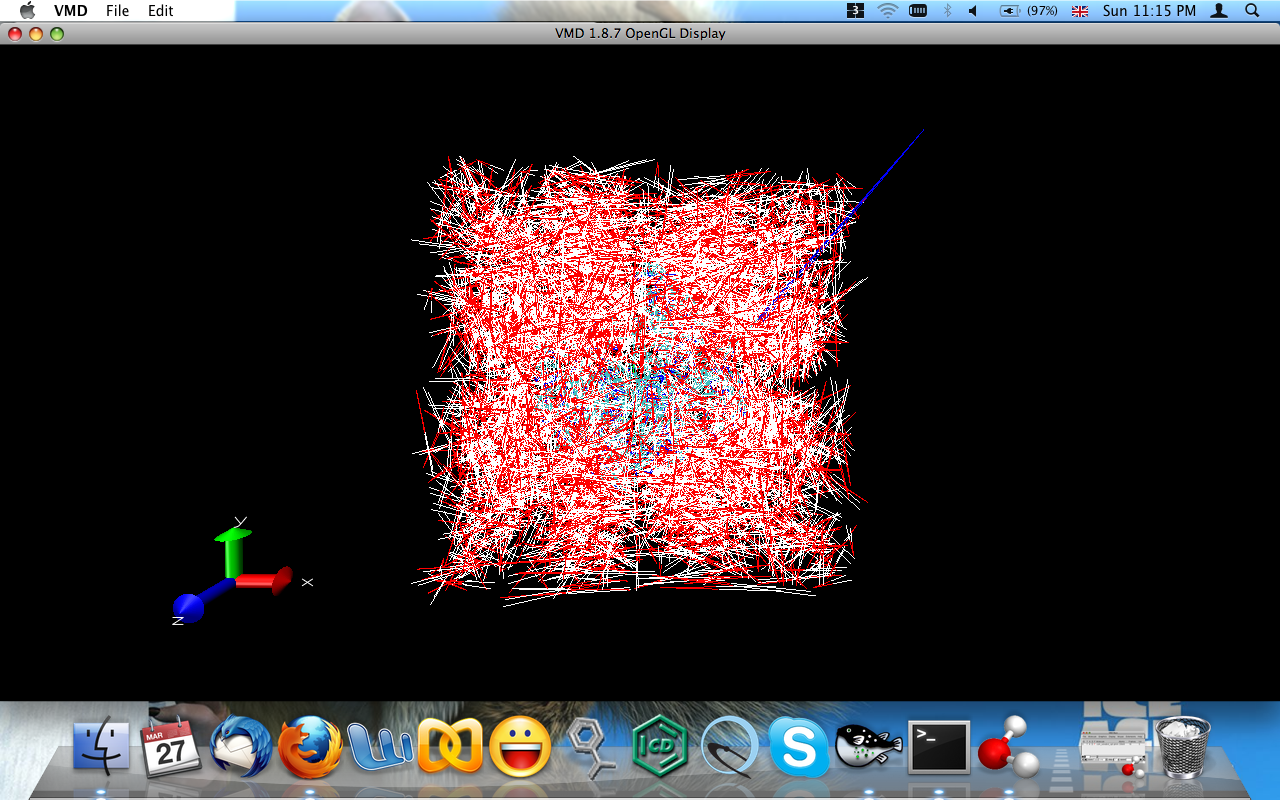
> > Looking at the image of your ligand, there is definitely something odd.
> > Are you using periodic boundaries when you load the ligand into VMD for
> > viewing? When I loaded the ligand from the files you sent into VMD using
> > Coordinates With Periodic Box info, it gives an image like you show.
> > When I load without Periodic Box info, it shows up just fine.
> >
> > I will see if I can replicate the NaN error under PBSA itself to see if
> > the source of the problem is there or something else.
> > -Wes
> >
> > > Dear Jason
> > > My input file consists of following parameters:
> > > Input file for running PB and GB
> > > &general
> > >
> > > startframe=1, endframe=50, interval=1,
> > >
> > > verbose=2, keep_files=0,
> > >
> > > /
> > > &gb
> > >
> > > igb=5, saltcon=0.150,
> > >
> > > /
> > > &pb
> > >
> > > istrng=0.15, fillratio=4.0
> > > /
> > >
> > > the command i used is:
> > > MMPBSA.py -O -i mmpbsa.in -o FINAL_RESULTS_MMPBSA.dat -sp
> > > amcligsolvated.prmtop -cp complex.prmtop -rp protein.prmtop -lp
> > > ligand.prmtop -y amcprod1.mdcrd
> > > command ran smooth no error appeared on terminal but when i checked
> > output
> > > file i came to know that NAN error in case of ligand occured because of
> > > which no energy difference calculated.
> > > Output was:
> > >
> > > | Run on Tue Sep 17 17:20:17 2013
> > > |
> > > |Input file:
> > > |--------------------------------------------------------------
> > > |Input file for running PB and GB in serial
> > > |&general
> > > | endframe=50, keep_files=2,
> > > |/
> > > |&gb
> > > | igb=2, saltcon=0.100,
> > > |/
> > > |&pb
> > > | istrng=0.100,
> > > |/
> > > | --------------------------------------------------------------
> > > |MMPBSA.py Version=12.0
> > > |Solvated complex topology file: amcligsolvated.prmtop
> > > |Complex topology file: amcliguUS.prmtop
> > > |Receptor topology file: pro.prmtop
> > > |Ligand topology file: unk.prmtop
> > > |Initial mdcrd(s): amcprod1.mdcrd
> > > |
> > > |Receptor mask: ":1-357"
> > > |Ligand mask: ":358"
> > > |Ligand residue name is "UNK"
> > > |
> > > |Calculations performed using 50 complex frames.
> > > |Poisson Boltzmann calculations performed using internal PBSA solver in
> > > mmpbsa_py_energy
> > > |
> > > |All units are reported in kcal/mole.
> > >
> >
> -------------------------------------------------------------------------------
> > >
> >
> -------------------------------------------------------------------------------
> > >
> > > GENERALIZED BORN:
> > >
> > > WARNING: INCONSISTENCIES EXIST WITHIN INTERNAL POTENTIAL
> > > TERMS. THE VALIDITY OF THESE RESULTS ARE HIGHLY QUESTIONABLE
> > > Complex:
> > > Energy Component Average Std. Dev. Std. Err.
> of
> > > Mean
> > >
> >
> -------------------------------------------------------------------------------
> > > BOND 1046.0599 25.0584
> > > 3.5438
> > > ANGLE 2825.6096 27.8852
> > > 3.9436
> > > DIHED 3867.0393 20.6518
> > > 2.9206
> > > VDWAALS -2766.8957 18.9666
> > > 2.6823
> > > EEL -22450.1018 52.8100
> > > 7.4685
> > > 1-4 VDW 1296.0772 16.0482
> > > 2.2696
> > > 1-4 EEL 14608.9084 34.7009
> > > 4.9074
> > > EGB -4636.7135 33.1652
> > > 4.6903
> > > ESURF 146.7058 1.1452
> > > 0.1620
> > >
> > > G gas -1573.3031 55.6723
> > > 7.8733
> > > G solv -4490.0077 33.3028
> > > 4.7097
> > >
> > > TOTAL -6063.3108 43.9275
> > > 6.2123
> > >
> > >
> > > Receptor:
> > > Energy Component Average Std. Dev. Std. Err.
> of
> > > Mean
> > >
> >
> -------------------------------------------------------------------------------
> > > BOND 1023.2391 25.1818
> > > 3.5612
> > > ANGLE 2758.3780 27.3729
> > > 3.8711
> > > DIHED 3808.4462 19.5218
> > > 2.7608
> > > VDWAALS -2656.1969 18.4136
> > > 2.6041
> > > EEL -22450.1018 52.8100
> > > 7.4685
> > > 1-4 VDW 1272.7137 15.8878
> > > 2.2469
> > > 1-4 EEL 14608.9084 34.7009
> > > 4.9074
> > > EGB -4662.4389 33.1432
> > > 4.6872
> > > ESURF 148.2532 1.1910
> > > 0.1684
> > >
> > > G gas -1634.6133 55.0064
> > > 7.7791
> > > G solv -4514.1857 33.2875
> > > 4.7076
> > >
> > > TOTAL -6148.7990 44.1153
> > > 6.2389
> > >
> > >
> > > Ligand:
> > > Energy Component Average Std. Dev. Std. Err.
> of
> > > Mean
> > >
> >
> -------------------------------------------------------------------------------
> > > BOND 22.8209 3.7323
> > > 0.5278
> > > ANGLE 67.2316 5.6725
> > > 0.8022
> > > DIHED 57.9771 3.3766
> > > 0.4775
> > > VDWAALS -21.1078 1.7341
> > > 0.2452
> > > 1-4 VDW 23.3635 2.0523
> > > 0.2902
> > > ESURF 8.4113 0.1439
> > > 0.0204
> > >
> > > G gas 150.2853 7.8027
> > > 1.1035
> > > G solv 8.4113 0.1439
> > > 0.0204
> > >
> > > TOTAL 158.6965 7.8116
> > > 1.1047
> > >
> > >
> > > Differences (Complex - Receptor - Ligand):
> > > Energy Component Average Std. Dev. Std. Err.
> of
> > > Mean
> > >
> >
> -------------------------------------------------------------------------------
> > > BOND -0.0000 0.0000
> > > 0.0000
> > > ANGLE -0.0000 0.0001
> > > 0.0000
> > > DIHED 0.6160 0.3232
> > > 0.0457
> > > VDWAALS -89.5910 3.1602
> > > 0.4469
> > > EEL 0.0000 0.0000
> > > 0.0000
> > > 1-4 VDW -0.0000 0.0001
> > > 0.0000
> > > 1-4 EEL 0.0000 0.0000
> > > 0.0000
> > > EGB 25.7254 1.2092
> > > 0.1710
> > > ESURF -9.9586 0.2770
> > > 0.0392
> > >
> > > DELTA G gas -88.9750 3.0895
> > > 0.4369
> > > DELTA G solv 15.7667 1.1381
> > > 0.1610
> > >
> > > DELTA TOTAL -73.2083 3.0542
> > > 0.4319
> > >
> > >
> > >
> >
> -------------------------------------------------------------------------------
> > >
> >
> -------------------------------------------------------------------------------
> > >
> > > POISSON BOLTZMANN:
> > >
> > > WARNING: INCONSISTENCIES EXIST WITHIN INTERNAL POTENTIAL
> > > TERMS. THE VALIDITY OF THESE RESULTS ARE HIGHLY QUESTIONABLE
> > > Complex:
> > > Energy Component Average Std. Dev. Std. Err.
> of
> > > Mean
> > >
> >
> -------------------------------------------------------------------------------
> > > BOND 1046.0599 25.0584
> > > 3.5438
> > > ANGLE 2825.6096 27.8852
> > > 3.9436
> > > DIHED 3867.0393 20.6518
> > > 2.9206
> > > VDWAALS -2766.8957 18.9666
> > > 2.6823
> > > EEL -22450.1018 52.8100
> > > 7.4685
> > > 1-4 VDW 1296.0772 16.0482
> > > 2.2696
> > > 1-4 EEL 14608.9084 34.7009
> > > 4.9074
> > > EPB -4507.0803 33.7185
> > > 4.7685
> > > ENPOLAR 2907.4367 7.0087
> > > 0.9912
> > >
> > > G gas -1573.3031 55.6723
> > > 7.8733
> > > G solv -1599.6436 35.2985
> > > 4.9920
> > >
> > > TOTAL -3172.9466 46.1397
> > > 6.5251
> > >
> > >
> > > Receptor:
> > > Energy Component Average Std. Dev. Std. Err.
> of
> > > Mean
> > >
> >
> -------------------------------------------------------------------------------
> > > BOND 1023.2391 25.1818
> > > 3.5612
> > > ANGLE 2758.3780 27.3729
> > > 3.8711
> > > DIHED 3808.4462 19.5218
> > > 2.7608
> > > VDWAALS -2656.1969 18.4136
> > > 2.6041
> > > EEL -22450.1018 52.8100
> > > 7.4685
> > > 1-4 VDW 1272.7137 15.8878
> > > 2.2469
> > > 1-4 EEL 14608.9084 34.7009
> > > 4.9074
> > > EPB -4549.0703 33.2881
> > > 4.7076
> > > ENPOLAR 2865.0984 7.0316
> > > 0.9944
> > >
> > > G gas -1634.6133 55.0064
> > > 7.7791
> > > G solv -1683.9719 34.8674
> > > 4.9310
> > >
> > > TOTAL -3318.5852 46.6278
> > > 6.5942
> > >
> > >
> > > Ligand:
> > > Energy Component Average Std. Dev. Std. Err.
> of
> > > Mean
> > >
> >
> -------------------------------------------------------------------------------
> > > BOND 22.8209 3.7323
> > > 0.5278
> > > ANGLE 67.2316 5.6725
> > > 0.8022
> > > DIHED 57.9771 3.3766
> > > 0.4775
> > > VDWAALS -21.1078 1.7341
> > > 0.2452
> > > 1-4 VDW 23.3635 2.0523
> > > 0.2902
> > > EPB nan nan
> > > nan
> > > ENPOLAR 97.8510 0.8688
> > > 0.1229
> > >
> > > G gas 150.2853 7.8027
> > > 1.1035
> > > G solv nan nan
> > > nan
> > >
> > > TOTAL nan nan
> > > nan
> > >
> > >
> > > Differences (Complex - Receptor - Ligand):
> > > Energy Component Average Std. Dev. Std. Err.
> of
> > > Mean
> > >
> >
> -------------------------------------------------------------------------------
> > > BOND -0.0000 0.0000
> > > 0.0000
> > > ANGLE -0.0000 0.0001
> > > 0.0000
> > > DIHED 0.6160 0.3232
> > > 0.0457
> > > VDWAALS -89.5910 3.1602
> > > 0.4469
> > > EEL 0.0000 0.0000
> > > 0.0000
> > > 1-4 VDW -0.0000 0.0001
> > > 0.0000
> > > 1-4 EEL 0.0000 0.0000
> > > 0.0000
> > > EPB nan nan
> > > nan
> > > ENPOLAR -55.5128 1.3871
> > > 0.1962
> > > EDISPER 0.0000 0.0000
> > > 0.0000
> > >
> > > DELTA G gas -88.9750 3.0895
> > > 0.4369
> > > DELTA G solv nan nan
> > > nan
> > >
> > > DELTA TOTAL nan nan
> > > nan
> > >
> > >
> > >
> >
> -------------------------------------------------------------------------------
> > >
> >
> -------------------------------------------------------------------------------
> > > when i visualized prmtop and MMBPSA.py generated mdcrds i found
> > > a distorted ligand. i also tried MMPBSA.pl but no results.
> > >
> > > kindly attached image of ligand hopefully these would be helpful for
> you
> > >
> > >
> > > Regards
> > >
> > >
> > >
> > >
> > >
> > > Sumra Wajid Abbasi
> > > PhD Scholar
> > > Computational Biology Lab, National Center for Bioinformatics
> > > Quaid-e-Azam University, Islamabad-45320 Pakistan.
> > >
> > >
> > > On Thu, Sep 19, 2013 at 3:41 PM, Jason Swails
> > > <jason.swails.gmail.com>wrote:
> > >
> > >> On Thu, Sep 19, 2013 at 4:15 AM, Sumra Wajid Abbasi 30-FBAS/MSBI/F09 <
> > >> sumra.msbi30.iiu.edu.pk> wrote:
> > >>
> > >> > Dear Jason
> > >> >
> > >> > NAN= No Atom Name
> > >> >
> > >>
> > >> Actually NaN means "Not a Number." [http://en.wikipedia.org/wiki/NaN
> ]
> > >> It is an almost universal sign of some type of problem (either user
> or
> > >> programmer).
> > >>
> > >> EPB/EGB = the electrostatic contribution to the solvation free energy
> > >> > calculated by PB or GB respectively
> > >> > ECAVITY = nonpolar contribution to the solvation free energy
> > >> calculated
> > >> by
> > >> > an empirical model
> > >> >
> > >>
> > >> You misunderstood me. I'm aware of what all of these terms mean.
> What
> > >> is
> > >> important when debugging is _which_ terms had NaN as their respective
> > >> values? Since the solvation free energy is NaN, that means either the
> > >> polar solvation term (EPB/EGB) or the non-polar solvation term
> (ECAVITY,
> > >> ESURF, EDISPER) must be NaN (or both). Knowing this gives clues about
> > >> where to look for problems. Of course you also omitted the G gas term
> > >> from
> > >> your email, so there could be problems there as well.
> > >>
> > >> I have visualized MMPBSA.py generated mdcrds using VMD...what i feel
> is
> > >> > there are abnormalities in ligand geometry and also few in
> receptor..
> > >> >
> > >>
> > >> This does not help, as it leaves me to "guess" what the
> 'abnormalities'
> > >> are
> > >> (and what you would consider 'normal'). For instance:
> > >>
> http://www.mybiosoftware.com/wp-content/uploads/2011/03/AmberTools.jpg
> > >> and http://archive.ambermd.org/201103/att-0730/Picture_2.png are two
> > >> different 'abnormalities' which have quite different root causes and
> > >> solutions. A picture is worth more than any number of words in this
> > >> case.
> > >>
> > >> As general advice when asking questions of this nature, you will
> receive
> > >> helpful answers far faster if you provide a good amount of details.
> > >> Show
> > >> the full output (not just lines you find troubling, because those are
> > >> often
> > >> not helpful by themselves). Show the exact commands that you used and
> > >> any
> > >> error messages or outputs that were produced. Also say how you tried
> to
> > >> fix or debug it, and what you learned (or think you learned) in the
> > >> process. Otherwise, the first few days will be spent trading emails
> of
> > >> this sort...
> > >>
> > >> HTH,
> > >> Jason
> > >>
> > >> --
> > >> Jason M. Swails
> > >> BioMaPS,
> > >> Rutgers University
> > >> Postdoctoral Researcher
> > >> _______________________________________________
> > >> AMBER mailing list
> > >> AMBER.ambermd.org
> > >> http://lists.ambermd.org/mailman/listinfo/amber
> > >>
> > > _______________________________________________
> > > AMBER mailing list
> > > AMBER.ambermd.org
> > > http://lists.ambermd.org/mailman/listinfo/amber
> > >
> >
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
> >
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
--
Jason M. Swails
BioMaPS,
Rutgers University
Postdoctoral Researcher
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Fri Sep 20 2013 - 04:00:03 PDT
{kind=link}
{kind=link}
