Date: Fri, 6 Sep 2013 20:03:05 +0530
Dear Roe
PFA!
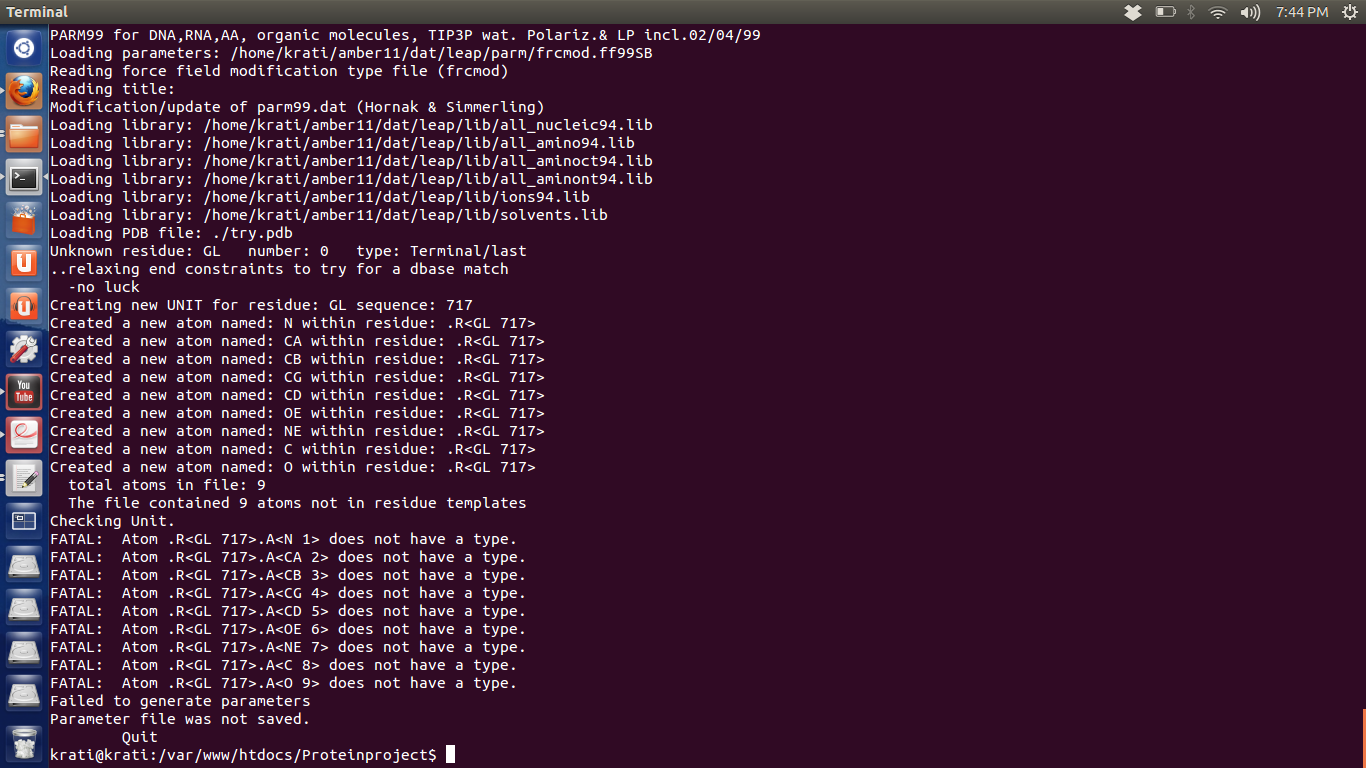
1. Error -try.png
2.
a. This is what in my actual file
ATOM 11531 CB GLNB 717 47.313 -3.313
-15.627
On running tleap, it shows error (try.png)
FATAL: Atom .R<GL 717>.A<CB 3> does not have a type.
b. When I reduce the gap between ATOM and 11532 (see below) I get
.top and .crd file
ATOM 11532 CG GLNB 717 47.822 -1.912 -15.233 (actual file-
Result in error)
ATOM 11533 CD GLNB 717 48.945 -1.897 -14.190(edit file- Result in
.top and .crd generation. Here it reads residue GLN instead just GL)
3. I tried using -aatm flag and without the same flag................ its
not helping me to solve this problem
Regards
Krati Sharma
On Fri, Sep 6, 2013 at 5:53 PM, David A Case <case.biomaps.rutgers.edu>wrote:
> On Fri, Sep 06, 2013, Krati Sharma wrote:
> >
> > I am generating the solvent file using command
> > ambpdb -aatm -p protein.top <protein_min.crd> protein_solvent.pdb
> >
> > When the protein is lengthy, its causing format problem during tleap run.
> >
> > If you see below the chain is ending at 2163 and the chain b is starting
> > from 2164 instead of 1.
>
> This is expected: the prmtop file that ambpdb is using numbers residues
> sequentially, not starting over again for each chain.
>
> >
> > ATOM 34303 C ARG 2163 5.387 47.227 -15.222 1.00
> > ATOM 34304 O ARG 2163 5.611 47.786 -14.144 1.00
> > ATOM 34305 OXT ARG 2163 5.440 47.901 -16.248 1.00
> > TER
> > ATOM 34306 N THR 2164 -15.132 51.856 34.718 1.00
> > ATOM 34307 H1 THR 2164 -14.494 51.092 34.890 1.00
> > ATOM 34308 H2 THR 2164 -15.462 51.795 33.765 1.00
> >
> > This cause error on running tlesp.If I reduce gap between ATOM and the
> > number 34303, tleap is reading full residue ARG otherwise it reads RG and
> > causing fatal error.
>
> I guess I can't see the problem: the "ARG" string for atom 34303 appears
> in columns 18-20, as it should. Can you give the exact error message from
> tleap? Can you be more specific about exactly what you did to "reduce the
> gap" between ATOM and the number? (what did the line originally look like?
> what did you change?)
>
> Is the result any better if you leave out the -aatm flag? You may be the
> only
> person who has used that option in years: it no longer is very useful
> now that the PDB has gone to the version 3 atom naming scheme. There
> could be
> some sort of bug with that flag.
>
> ...dac
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- *Krati Sharma Research scholar UNB (Scfbio*-*IIT) *
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: try.png)
