Date: Wed, 14 Aug 2013 10:38:18 -0500
Thank you Dr. Case. The desc command "desc MG2.1.MG" worked and I can see the charge on MG is 2.
So, what might be the reason for the metals to start to move away?
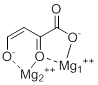
Mg2+ ion 1 is octahedrally coordinated by His, Asp, Glu, Ile, and 2 oxygen atoms from the inhibitor and Mg2+ ion 2 is tetrahedrally coordinated by Glu, Asp, and 2 oxygen atoms from the inhibitor. In the following pictorial representation, the planar oxygen atoms of the ligand are given.
[cid:image004.png.01CE98D8.D60FA330]
I tried to create distance restraints and I got the following error and the makeDIST_RST gives error as map.DG-AMBER do not recognize MG. In one similar post (year 2005), you suggested to use raw "Sander" format. However, I couldn't figure out how to implement this.
I tried to impose restraints on one of the "O" atoms, but didn't seem to work. I tried restraint_wt from 1 to 10.
restraint_wt=2.0,
restraintmask=':199.O28'
What do you suggest?
I am not sure if I can impose position restraints on Mg, as Mg2+ ion1 coordinates with a Glu, which is Asp in one mutant. I intend to study the effect of this mutation.
Thank you for your time and any suggestion you may offer in this regard.
Thanks and regards,
Latha.
________________________________
Email Disclaimer: www.stjude.org/emaildisclaimer
Consultation Disclaimer: www.stjude.org/consultationdisclaimer
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- application/octet-stream attachment: oledata.mso
- application/octet-stream attachment: image003.emz
(image/png attachment: image004.png)
