Date: Sun, 24 Mar 2013 11:23:15 +0530
Dear Dr. Jason and Amber people,
Thanks for your suggestion given here,
http://archive.ambermd.org/201303/0168.html and associated threads.
I have followed your suggestions and found that when I added charges of CYP
and HEM units, they summed up to exactly 2.000. CYP is a modified unit for
CYS435 residue.
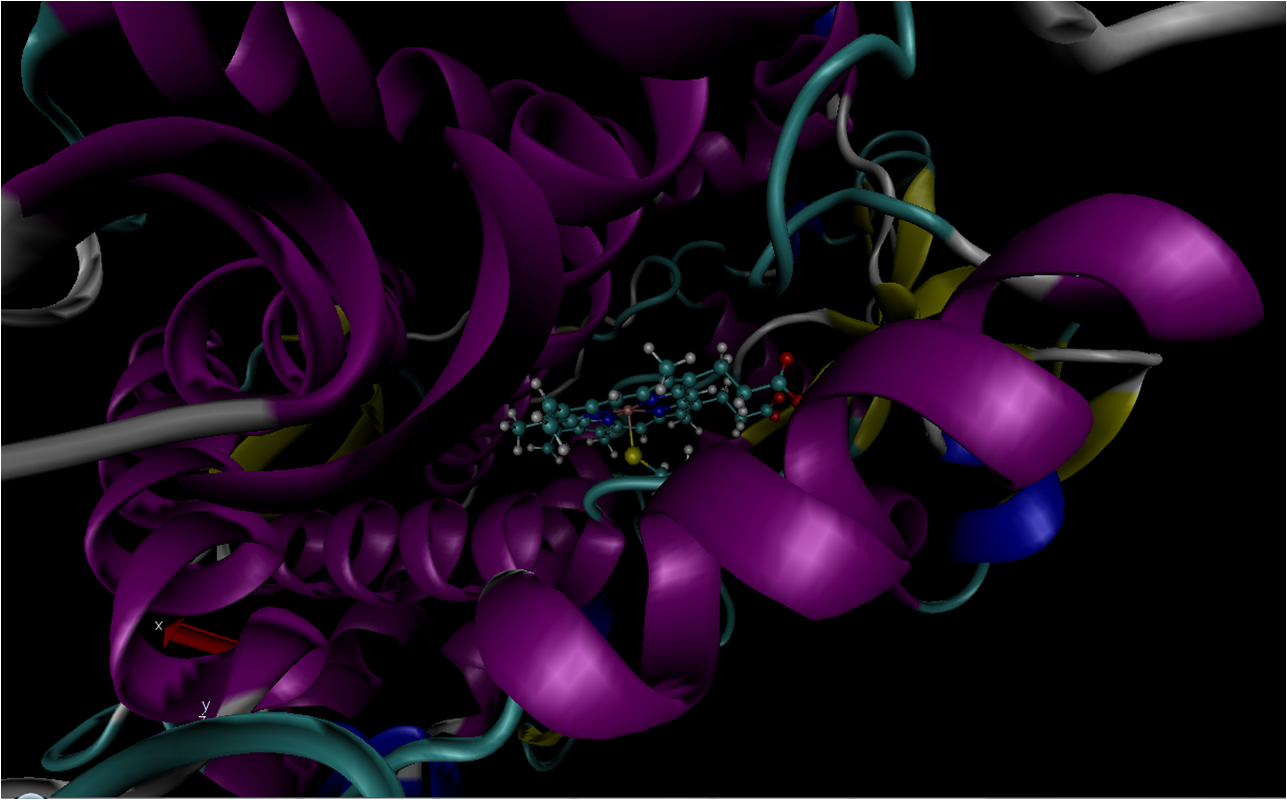
Then I have connected the S and Fe atoms with the command "bond
2vn0.435.SG<http://2vn0.435.sg/> 2vn0.492.FE"
and it seems to have worked as seen in the snapshot of prmtop and inpcrd
loaded in VMD (attached).
There was "one sided connection" warning for which a search suggested that
I should add TER card before and after CYP, but since it is part of the
protein chain, I did not add any TER card in the pdb.
http://www.rosswalker.co.uk/tutorials/amber_workshop/Tutorial_one/section2.htm
Please find time to suggest if the input files that I have generated here
are compatible for starting a meaningful MD simulation or there
are warnings/errors which I should be taking care of before starting any MD
on these prmtop and inpcrd files.
Thanks again for your valuable inputs.
gm.linux-rfml:~/Documents/MD/CYP450/TGZ_2vn0> tleap -s -f leap_TGZ.in
-I: Adding /usr/local/amber12/dat/leap/prep to search path.
-I: Adding /usr/local/amber12/dat/leap/lib to search path.
-I: Adding /usr/local/amber12/dat/leap/parm to search path.
-I: Adding /usr/local/amber12/dat/leap/cmd to search path.
-s: Ignoring startup file: leaprc
-f: Source leap_TGZ.in.
Welcome to LEaP!
Sourcing: ./leap_TGZ.in
----- Source: /usr/local/amber12/dat/leap/cmd/leaprc.ff99SB
----- Source of /usr/local/amber12/dat/leap/cmd/leaprc.ff99SB done
Log file: ./leap.log
Loading parameters: /usr/local/amber12/dat/leap/parm/parm99.dat
Reading title:
PARM99 for DNA,RNA,AA, organic molecules, TIP3P wat. Polariz.& LP
incl.02/04/99
Loading parameters: /usr/local/amber12/dat/leap/parm/frcmod.ff99SB
Reading force field modification type file (frcmod)
Reading title:
Modification/update of parm99.dat (Hornak & Simmerling)
Loading library: /usr/local/amber12/dat/leap/lib/all_nucleic94.lib
Loading library: /usr/local/amber12/dat/leap/lib/all_amino94.lib
Loading library: /usr/local/amber12/dat/leap/lib/all_aminoct94.lib
Loading library: /usr/local/amber12/dat/leap/lib/all_aminont94.lib
Loading library: /usr/local/amber12/dat/leap/lib/ions94.lib
Loading library: /usr/local/amber12/dat/leap/lib/solvents.lib
----- Source: /usr/local/amber12/dat/leap/cmd/leaprc.gaff
----- Source of /usr/local/amber12/dat/leap/cmd/leaprc.gaff done
Log file: ./leap.log
Loading parameters: /usr/local/amber12/dat/leap/parm/gaff.dat
Reading title:
AMBER General Force Field for organic molecules (Version 1.4, March 2010)
add. info. at the end
Loading Mol2 file: ./CYP.mol2
Reading MOLECULE named CYP-IC6
Loading parameters: ./Heme.frcmod
Reading force field modification type file (frcmod)
Reading title:
remark goes here
Loading Mol2 file: ./Heme.mol2
Reading MOLECULE named HEM
Loading PDB file: ./2VN0_heme2.pdb
One sided connection. Residue: CYP-IC6 missing connect0 atom.
One sided connection. Residue: default_name missing connect1 atom.
Added missing heavy atom: .R<CHIE 491>.A<OXT 18>
+Currently only Sp3-Sp3/Sp3-Sp2/Sp2-Sp2 are supported
+---Tried to superimpose torsions for: *-NB-FE-*
+--- With Sp2 - Sp0
+--- Sp0 probably means a new atom type is involved
+--- which needs to be added via addAtomTypes
+Currently only Sp3-Sp3/Sp3-Sp2/Sp2-Sp2 are supported
+---Tried to superimpose torsions for: *-NA-FE-*
+--- With Sp2 - Sp0
+--- Sp0 probably means a new atom type is involved
+--- which needs to be added via addAtomTypes
+Currently only Sp3-Sp3/Sp3-Sp2/Sp2-Sp2 are supported
+---Tried to superimpose torsions for: *-ND-FE-*
+--- With Sp2 - Sp0
+--- Sp0 probably means a new atom type is involved
+--- which needs to be added via addAtomTypes
+Currently only Sp3-Sp3/Sp3-Sp2/Sp2-Sp2 are supported
+---Tried to superimpose torsions for: *-NC-FE-*
+--- With Sp2 - Sp0
+--- Sp0 probably means a new atom type is involved
+--- which needs to be added via addAtomTypes
total atoms in file: 7491
Leap added 11 missing atoms according to residue templates:
1 Heavy
10 H / lone pairs
> bond 2vn0.435.SG <http://2vn0.435.sg/> 2vn0.492.FE
> check CYP
Checking 'CYP'....
ERROR: The unperturbed charge of the unit: -0.402400 is not integral.
WARNING: The unperturbed charge of the unit: -0.402400 is not zero.
Checking parameters for unit 'CYP'.
Checking for bond parameters.
Checking for angle parameters.
check: Errors: 1 Warnings: 1
> check HEM
Checking 'HEM'....
ERROR: The unperturbed charge of the unit: -1.597600 is not integral.
WARNING: The unperturbed charge of the unit: -1.597600 is not zero.
Checking parameters for unit 'HEM'.
Checking for bond parameters.
Checking for angle parameters.
check: Errors: 1 Warnings: 1
> charge 2vn0
Total unperturbed charge: 4.000000
Total perturbed charge: 4.000000
> addions 2vn0 Cl- 0
4 Cl- ions required to neutralize.
Adding 4 counter ions to "2vn0" using 1A grid
Grid extends from solute vdw + 2.47 to 8.47
Resolution: 1.00 Angstrom.
grid build: 1 sec
(no solvent present)
Calculating grid charges
charges: 10 sec
Placed Cl- in 2vn0 at (64.02, 27.80, 6.36).
Placed Cl- in 2vn0 at (33.02, 6.80, -17.64).
Placed Cl- in 2vn0 at (71.02, 17.80, -17.64).
Placed Cl- in 2vn0 at (43.02, -2.20, -36.64).
Done adding ions.
> solvateoct 2vn0 TIP3PBOX 8.0
Scaling up box by a factor of 1.346722 to meet diagonal cut criterion
Solute vdw bounding box: 67.160 51.455 74.221
Total bounding box for atom centers: 95.768 95.768 95.768
(box expansion for 'iso' is 41.6%)
Solvent unit box: 18.774 18.774 18.774
Volume: 453071.057 A^3 (oct)
Total mass 244848.466 amu, Density 0.897 g/cc
Added 10626 residues.
> saveamberparm 2vn0 2vn0_apo.prmtop 2vn0_apo.inpcrd
Checking Unit.
Building topology.
Building atom parameters.
Building bond parameters.
Building angle parameters.
Building proper torsion parameters.
Building improper torsion parameters.
total 1473 improper torsions applied
Building H-Bond parameters.
Not Marking per-residue atom chain types.
Marking per-residue atom chain types.
(Residues lacking connect0/connect1 -
these don't have chain types marked:
res total affected
CHIE 1
CYP 1
HEM 1
NLYS 1
WAT 10626
)
(no restraints)
>
> check 2vn0
Checking '2vn0'....
Warning: Close contact of 1.430419 angstroms between .R<NLYS 28>.A<HD3 15>
and .R<NLYS 28>.A<H3 4>
Warning: Close contact of 1.493880 angstroms between .R<NLYS 28>.A<HD3 15>
and .R<NLYS 28>.A<H1 2>
Warning: Close contact of 0.109243 angstroms between .R<NLYS 28>.A<H3 4>
and .R<NLYS 28>.A<H1 2>
Warning: Close contact of 1.313180 angstroms between .R<ILE 38>.A<HG23 10>
and .R<ILE 39>.A<HG23 10>
Warning: Close contact of 1.454352 angstroms between .R<LYS 118>.A<HZ1 18>
and .R<LYS 279>.A<HD3 13>
Warning: Close contact of 1.314590 angstroms between .R<MET 136>.A<HE3 15>
and .R<ASP 262>.A<HA 4>
Warning: Close contact of 1.421075 angstroms between .R<ILE 141>.A<HA 4>
and .R<ARG 144>.A<HD2 12>
Warning: Close contact of 1.299580 angstroms between .R<PRO 166>.A<HB2 9>
and .R<LEU 170>.A<HD22 16>
Warning: Close contact of 1.401001 angstroms between .R<LEU 170>.A<HD21 15>
and .R<LEU 310>.A<HD12 12>
Warning: Close contact of 1.412214 angstroms between .R<ALA 173>.A<HB3 8>
and .R<PRO 174>.A<HD2 3>
Warning: Close contact of 1.461887 angstroms between .R<ARG 261>.A<HE 15>
and .R<ASP 265>.A<OD2 10>
Warning: Close contact of 1.172415 angstroms between .R<ILE 331>.A<HD13 17>
and .R<PRO 337>.A<HB3 10>
Warning: Close contact of 1.024731 angstroms between .R<LYS 415>.A<HD2 12>
and .R<LYS 415>.A<H 2>
Warning: Close contact of 1.332051 angstroms between .R<ILE 434>.A<C 18>
and .R<CYP 435>.A<N 1>
Warning: Close contact of 1.326672 angstroms between .R<CYP 435>.A<C 9> and
.R<ALA 436>.A<N 1>
Checking parameters for unit '2vn0'.
Checking for bond parameters.
Checking for angle parameters.
check: Warnings: 15
Unit is OK.
-- With regards Vaibhav A. Dixit Ph.D. Department of Medicinal Chemistry Natl. Inst. Pharm. Edu. & Res. (NIPER) Sector 67, Phase X, S.A.S. Nagar (Mohali) Punjab -160 062 INDIA Phone (Mobile): +919915214408, +91-7709129400. E-mail: vaibhavadixit.gmail.com www.niper.nic.in
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 2vn0_heme.png)
