Date: Fri, 1 Feb 2013 02:16:09 +0000 (UTC)
Hi all,
I am interested in running a minimization on a lengthy peptide. I would like to use a cubic solvation box for periodicity. I have been encountering some problems as I change the box length. At 20 ang, the box seems to small, and I am worried that conformational changes could lead to interactions with the neighboring replica. If I make the box bigger (i.e. box length = 60 ang), the boxes of water look like they are stacking on top of each other.
I am running the tleap script from a python subprocess. Originally, I speculated that this could lead to some problems. However, I have manually solvated this system, and the same thing happens. My tleap script is below:
#!/bin/bash
source leaprc.ff12SB
l100 = loadpdb lambdaN_100.pdb
solvatebox l100 TIP3PBOX 20
savepdb l100 wb_lambdaN_100.pdb
quit
!
To sum, I am searching for a way to solvate a long peptide.
Thanks,
Joe Passman
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
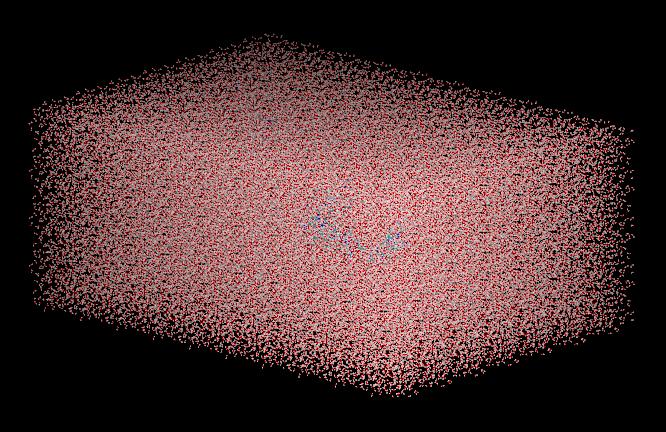
(image/jpeg attachment: BoxLength20.jpg)
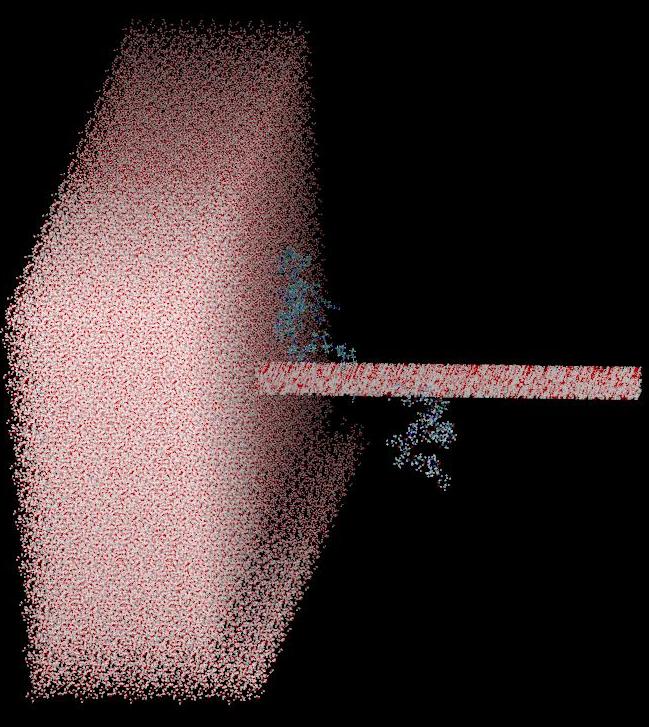
(image/jpeg attachment: BoxLength60.jpg)
