Date: Wed, 7 Nov 2012 13:20:37 -0700
Hi,
Based on the files you gave me it doesn't look like there is a problem
with the autoimage command. I think the issue here may be your atom
masks. Your system is made up of 6 large (~3300 atom) molecules, ions,
and solvent. The first plot you show (nwrms) is generated from the
command 'rms mass out nwrms.out :1-600' - however, according to the
parmtop you sent me this spans several monomers as well as some ions
(here is output for your system from cpptraj 'parmmolinfo'):
MOLECULES:
Molecule 1, 3381 atoms, first residue GLY_1
Molecule 2, 1 atoms, first residue K+ _224
Molecule 3, 1 atoms, first residue K+ _225
Molecule 4, 1 atoms, first residue Cl-_226
Molecule 5, 3398 atoms, first residue PHE_227
Molecule 6, 1 atoms, first residue K+ _450
Molecule 7, 1 atoms, first residue Cl-_451
Molecule 8, 3365 atoms, first residue TYR_452
Molecule 9, 1 atoms, first residue K+ _674
Molecule 10, 1 atoms, first residue K+ _675
Molecule 11, 1 atoms, first residue Cl-_676
Molecule 12, 1 atoms, first residue Ca2_677
Molecule 13, 3405 atoms, first residue GLY_678
...
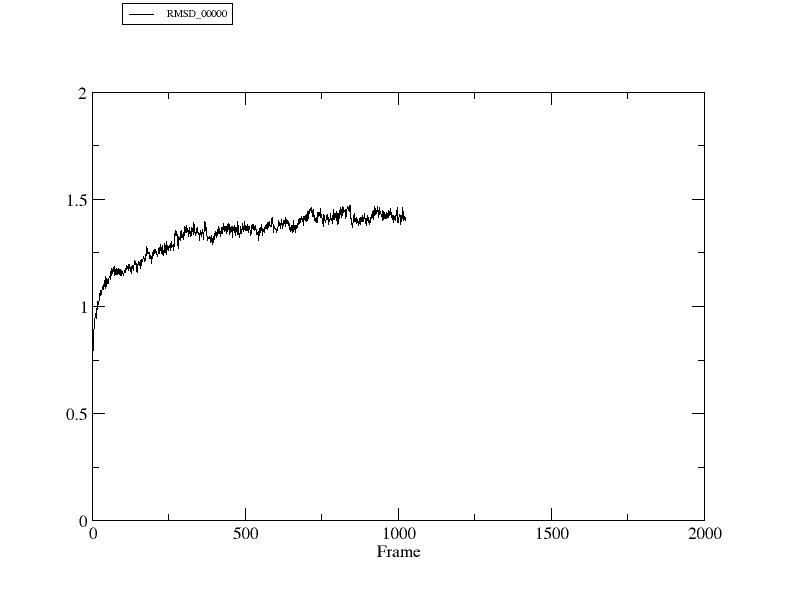
The movement of ions is probably what is causing the little spikes in
your RMS plot (indeed, if I change it to include the ions I get those
spikes as well). If I generate an RMSD of all the monomers (no ions or
solvent) to the first frame of the trajectory it looks relatively
smooth (see attached plot). This was generated from:
parm hex.parm7
trajin ...
strip :WAT,K+,Cl-
rms first out rmsd.agr
One thing to be careful of is after a 'strip' command your
atoms/residues become renumbered, so in this case after the strip your
second monomer would start at residue 224 instead of 227 (since the
ions that were in-between are now stripped).
The second plot you show seems completely normal. If you visualize the
trajectory you can see that the spikes correspond to normal changes in
the ARG conformation.
Hope this helped.
-Dan
On Tue, Nov 6, 2012 at 3:52 PM, Brown, Kyle <kyle.l.brown.vanderbilt.edu> wrote:
> Hi Guys,
> Thank you so much for your help. I am sorry to report that autoimage in
> ccptraj was not a panacea for my IMAGE conundrum. Attached you can view
> the rms for the entire system and for residue 165. There still seems to
> be something amiss. My input files was as follows:
>
> trajin ../100.mdcrd.gz
>
> .
> .
> [sic]
> .
> .
> trajin ../128.mdcrd.gz
> trajin ../129.mdcrd.gz
> trajin ../130.mdcrd.gz
> reference min.rst
> autoimage
> rms reference out R165rms.out :165
> rms mass out nwrms.out :1-600
> atomicfluct out back.txt .C,CA,N byres bfactor
> strip :WAT
> average ccp_avg.pdb pdb
>
> Thank you in advance for your suggestions.
> KB
>
>
> On 11/6/12 2:36 PM, "Daniel Roe" <daniel.r.roe.gmail.com> wrote:
>
>>Hi,
>>
>>Just to add on to what Dave said, if you do use the autoimage command
>>that should be the only command you need, so your script would become:
>>
>>reference min.rst
>>autoimage
>>rms reference out R165rms.out time 0.01 :165 name R165rms
>>rms mass out nwrms.out time 0.01 :1-600 name mwrms
>>atomicfluct out back.txt .C,CA,N byres bfactor
>>strip :WAT
>>average avg.pdb pdb
>>
>>I recommend you try it on a subset of your trajectory first (ideally a
>>small range of frames where you know imaging is a problem). Let me
>>know if you run into problems.
>>
>>-Dan
>>
>>--
>>-------------------------
>>Daniel R. Roe, PhD
>>Department of Medicinal Chemistry
>>University of Utah
>>30 South 2000 East, Room 201
>>Salt Lake City, UT 84112-5820
>>http://home.chpc.utah.edu/~cheatham/
>>(801) 587-9652
>>(801) 585-9119 (Fax)
>>
>>_______________________________________________
>>AMBER mailing list
>>AMBER.ambermd.org
>>http://lists.ambermd.org/mailman/listinfo/amber
>>
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- ------------------------- Daniel R. Roe, PhD Department of Medicinal Chemistry University of Utah 30 South 2000 East, Room 201 Salt Lake City, UT 84112-5820 http://home.chpc.utah.edu/~cheatham/ (801) 587-9652 (801) 585-9119 (Fax)
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: rmsd.jpg)
