From: Brown, Kyle <kyle.l.brown.Vanderbilt.Edu>
Date: Tue, 6 Nov 2012 19:57:05 +0000
Greetings AMBER friends,
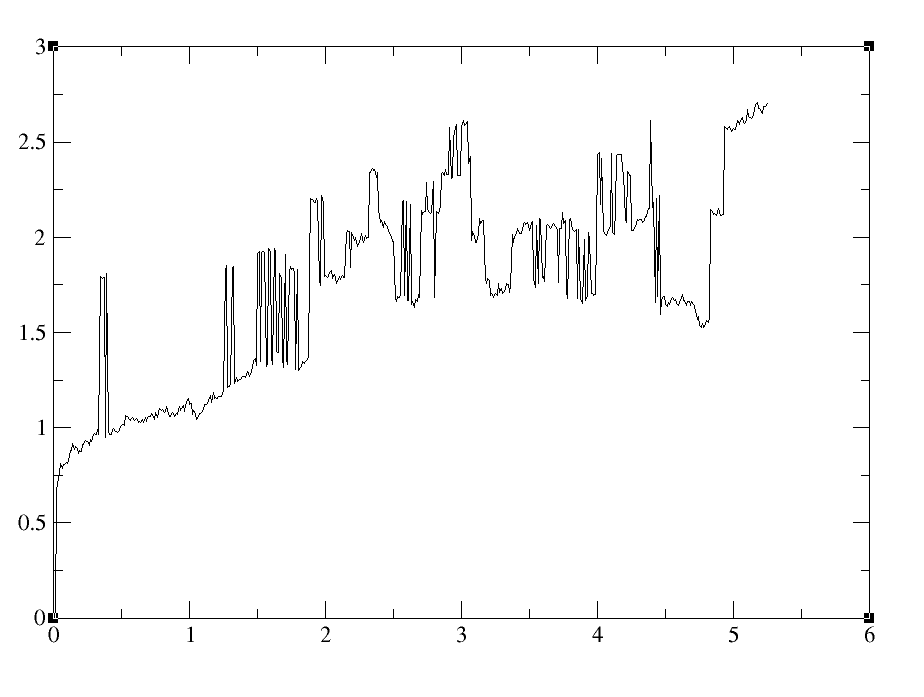
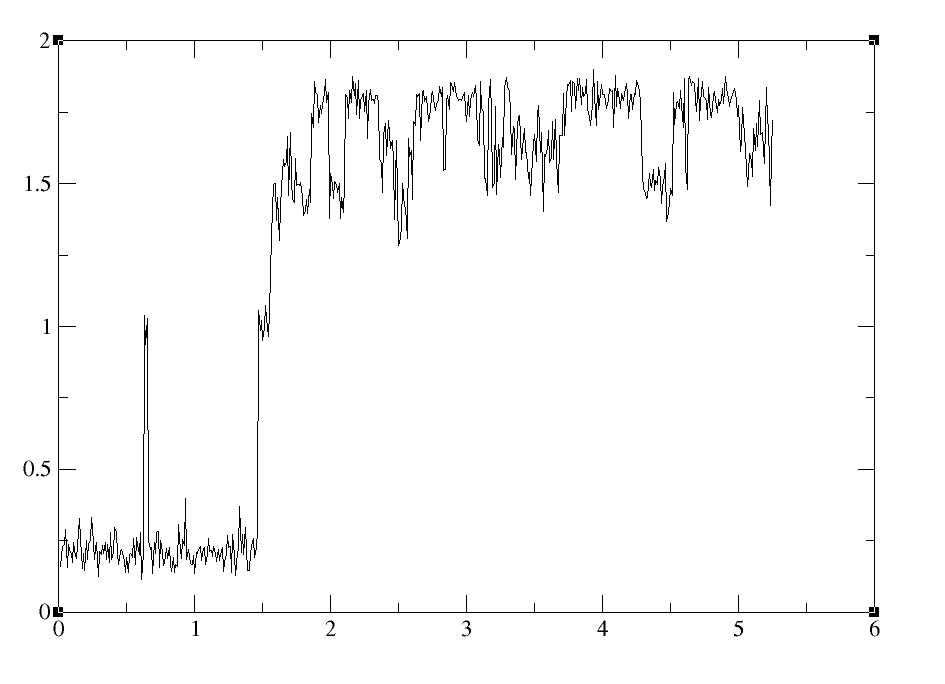
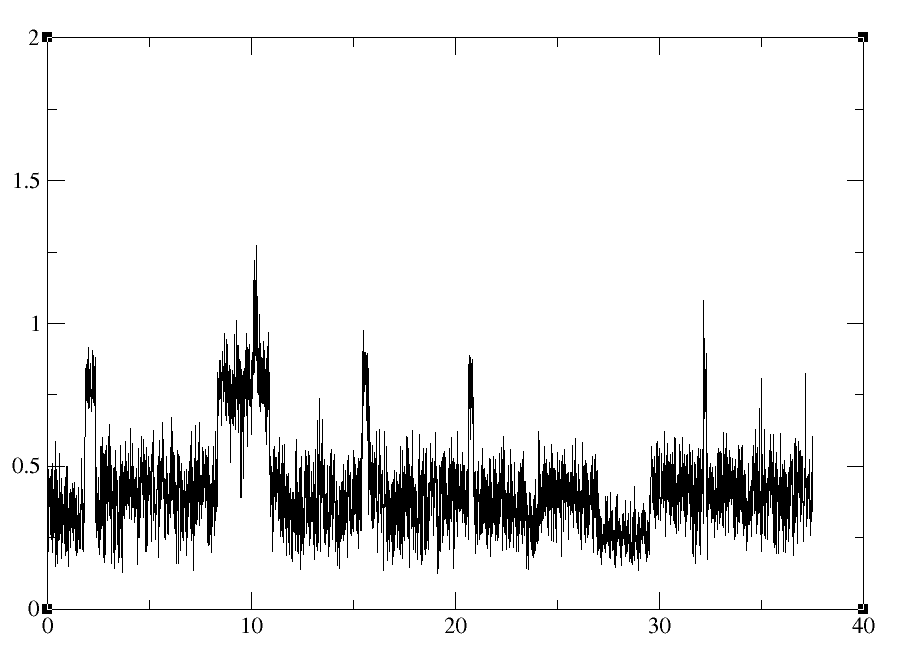
I am having problems imaging trajectories after lengthy simulations with IWRAP=1. I see this is a common problem judging by the posts on the mail server. I found a response by TEC to a previous post to be quite informative (http://archive.ambermd.org/201105/0225.html). Similar to the originator of this post, I have a large system composed of 6 monomers. During the course of the trajectory, one of the monomers is wrapped to the opposite corner of the periodic box. When performing the "two stage" centering/imaging with ptraj as recommended in the post, I get what seem to be unreasonable results as typified in a mass weighted rms of the system (see attached figure 1 for a small sample). I see bfactors in the range of 40-50A. For more specific examples see attached rms values for residues R165 and R400. A typical ptraj script is as follows assuming 100 residues per monomer:
center :1-100 mass origin
image origin center
center :1-600 mass origin
image origin center
reference min.rst
rms reference out R165rms.out time 0.01 :165 name R165rms
rms mass out nwrms.out time 0.01 :1-600 name mwrms
atomicfluct out back.txt .C,CA,N byres bfactor
strip :WAT
average avg.pdb pdb
Removing the "center" command as suggest in the referenced post results in two halves of the system being imaged to opposite sides of the periodic box. Performing a 6 stage center/imaging process for each monomer is equally unfruitful. I have attempted various iterations of center/imaging of specific sections of the system before others and in different orders, +/- "mass", +/- "origin"…. all to no avail. At this point I would certainly welcome any advice on the matter.
KB
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Tue Nov 06 2012 - 12:00:04 PST