Date: Sat, 14 Jul 2012 00:57:58 -0700 (PDT)
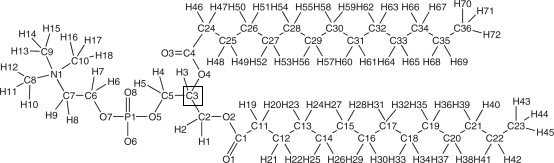
Dear Profs. Vlad, Ross, Gould and Benjamin, Thanks for your replies and as this discussion was spreading much, I am trying to put all together and humbly await for further helpful replies/suggestions: 1) *** Prof. Ross and Benjamin, >> It should work with all those offered by the Charmm GUI. What do you need specifically? charmmlipid2amber.x does not show DMPC (as it shows this message:: Currently supported lipids : DPPC DPPE DPPS DPPG DPPA DOPC DOPE DOPS DOPG DOPA POPC POPE POPS POPG POPA CHL1) 2) *** Prof./Dr. Benjamin, >>I assume you're talking about the stereogenic center of the phospholipid To avoid further confusion, I have attached the image (source: JCC,29(1), 24-37). I was discussing about the boxed carbon C3 (it is DMPC molecule). No other chiral centre I can see (please correct me). It should be 'R' as what CHARMM GUI provides us but 'S' was used in mentioned paper. Is there any special reason to use S and its validation with GAFF? 3) *** Prof. Junmie Wang (gaff developer), Gould, >>The P4 is correct, the phosphorous has three different substituents attached, the two oxygens which are >> attached to the phosphorous are equivalent. I am confused about the term "equivalent" and new for me. If it is the case then atom type like p5 (which I think was suitable instead of p4) will loose its significance to exist even!. lets say what gaff provides "p5 Phosphate with four connected atoms, such as O=P(OH)3". here also one can see three OHs being "equivalent", then can I say that atom type p2 is also OK or something like that. thanks and regards, ________________________________ From: Benjamin Madej <bmadej.ucsd.edu> To: 'AMBER Mailing List' <amber.ambermd.org> Sent: Saturday, July 14, 2012 12:44 AM Subject: Re: [AMBER] lipid params 2) Secondly the paper uses and validate many things with "S" configuration DMPC bilayer (the single residue is in "S" configuration, as provided in Richard's Bryce website). So can I assume these validations remain same for "R" configurations as DMPC lipid is R (I dont have reference for that though, but what CHARMM-GUI provides and other lipids are in R form only.). So is there any special reason 'S' lipid was used in such study? Anyhow I can use params for R also. I assume you're talking about the stereogenic center of the phospholipid (the sn-1 carbon) here. I believe for glycerophospholipids this is usually in the R configuration (J. A. Smith Organic Chemistry). As a side note, it is important to note that phosphatidylglycerol has another chiral center, but only the S configuration occurs in nature (K. K. Eklund et al. Biochemistry 1987). 3) Few lipids are there now in lipid11 (that charmmlipid2amber.x uses). As far I was following recent archive mails AMBER will provide params which can be used without surface tension once it is published. So the params in lipid11 will change after it is published or these are same params which can be used without surface tension? Actually, many of the CHARMM-GUI lipids are supported with Lipid11. Lipid11 is a modular force field, so it supports many different combinations of head groups and tail groups. See the Amber manual for a current list. However, myristoyl was not included in the first version of Lipid11. Because we are also interested in myristoyl, I am working on adding myristoyl to the parameter set. Currently, the Lipid11 parameters require a surface tension ensemble. Running bilayer systems with Lipid11 and a tensionless ensemble does not fully capture several experimental structural and dynamical properties. The surface tension ensemble with this parameter set approximates the bilayer behavior better for now. However, we are continuing to refine and test parameters that will be published in the future. These have not been released yet, but will be the next version of the Amber lipid force field included in AmberTools. Ben Madej Walker Molecular Dynamics Lab _______________________________________________ AMBER mailing list AMBER.ambermd.org http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: dmpc.jpg)
