Date: Thu, 10 May 2012 09:40:15 +0200
enclosed this email, you can find two screenshot for it..... as we can
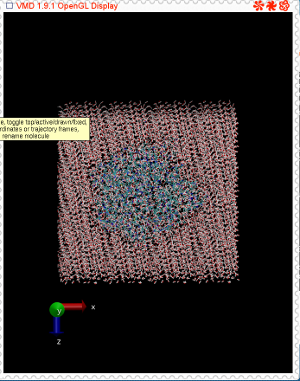
see, the density of solvent molecules in the second figure is rather low
and it spread everywhere..... I am very curious bout this simulations
has PBC.....
thank you very much
On 05/10/2012 09:33 AM, Albert wrote:
> Hi Marc:
>
> I am not sure about this, because each time after NVT, I found that
> my water molecules doesn't stay in the same box, it diffused......
> here is my NVT input file:
>
>
> heating gradually over 300ps
> &cntrl
> imin=0,irest=0,ntx=1,
> nstlim=300000,dt=0.001,
> ntc=2,ntf=2,
> cut=10.0, ntb=1,
> ntpr=500, ntwx=500,
> ntt=3, gamma_ln=2.0,
> tempi=0.0, temp0=310.0,
> restraintmask=':1-677 & !.H=',
> restraint_wt=10.0,
> nmropt=1
> /
> &wt TYPE='TEMP0', istep1=0, istep2=300000,
> value1=0.1, value2=310.0, /
> &wt TYPE='END' /
>
>
>
> On 05/10/2012 09:30 AM, Marc van der Kamp wrote:
>> Sounds like something for the VMD mailing list...
>> Marc
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot.png)

(image/png attachment: Screenshot-1.png)
