Date: Tue, 14 Feb 2012 16:40:06 +0100
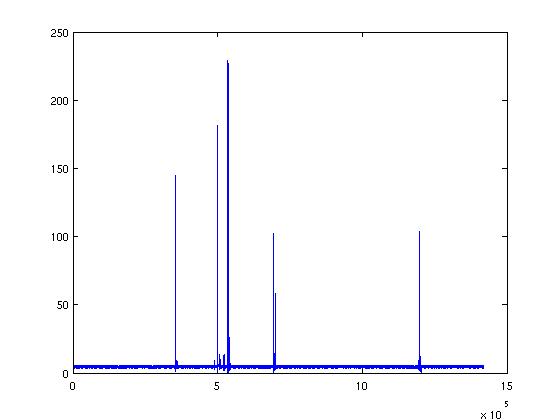
I'm calculating the distance from two atoms, the trajectory is without
solvent and I use netcdf files as input.
The figure show the distance of two atoms during the trajectory.
Andrea
On 02/14/2012 04:24 PM, Daniel Roe wrote:
> Hi,
>
> On Tue, Feb 14, 2012 at 9:54 AM, Giachetti Andrea - CERM
> <giachetti.cerm.unifi.it> wrote:
>> Hi all, I'm trying to extract coordinates from amber trj trajectory
>> file, but there are some steps with discontinuities (jumps). Could it be
>> related to periodic boundary conditions ?
> What exactly do you mean by discontinuities? Are they in something you
> are calculating (i.e. distance, RMSD, etc) or in the physical
> coordinates of the molecule (or both?). It would probably be useful to
> know something about the original trajectory (does it have solvent,
> PBC, etc).
>
> -Dan
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: distance.jpg)
