Date: Fri, 27 Jan 2012 16:12:33 +0300
Dear Amber community!
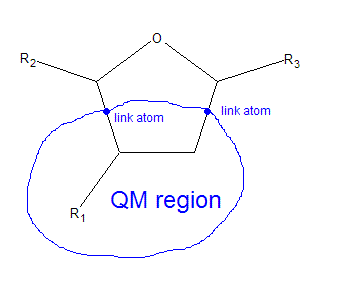
I want to split a cyclic molecule into quantum mechanics (QM) and
molecular mechanics (MM) parts during QM/MM MD simulation (with the
use of semiempirical methods incorporated into Amber10 Sander module)
as depicted in the attached file. But I am not shure that such a
partitioning is acceptable. Are there any known problems related to
splitting a cycle into QM and MM regions?
Thanks in advance and excuse me for this off-topic question!
-- Dmitry Nilov Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University web: http://enzyme.fbb.msu.ru/
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/gif attachment: qmmm.GIF)
