Date: Wed, 16 Nov 2011 12:09:34 -0600
Dear Amber Users,
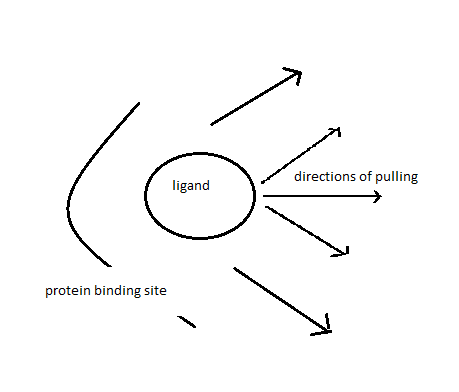
I want to pull the ligand out of binding pocket in various
directions/orientations. I have attached a cartoon representation for the
same.
If I'm using nmropt= 1 and doing umbrella sampling, then I can use the
distance restraints for COM distance between the C-alpha of the binding
site and ligand but how can I achieve directional pulling to generate
snapshots for umbrella sampling along that direction of pulling?
I have looked at the ncsu_smd section of chapter 4 where we can define the
path =x,x,x or define particularly the y-direction pulling. If I use the
z-direction pulling, then I can orient my protein-ligand complex along the
z-directions in various ways and achieve it. But is this idea robust?
The other way I can think of is using three C-alpha atoms of protein
binding site and one atom of ligand and get a dihedral angle for the four
atoms and restrain it. But adding additional restraints may effect the
calculated free energy. Or am I wrong?
Any suggestions will be helpful.
Thanks for your time.
Regards
Sai[image: ligand-pulling.png]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: ligand-pulling.png)
