Date: Wed, 20 Jul 2011 11:57:58 -0400
Hi all,
I have a question regarding the distribution of urea and water molecules
when a protein is solvated in a 8M urea box using tleap.
Here is the my input:
tleap -s -f leaprc.ff99SB
> loadAMBERparams frcmod.urea
Loading parameters: /Users/ndang/amber11/dat/leap/parm/frcmod.urea
Reading force field modification type file (frcmod)
Reading title:
Parameters of N - C - N angle and corresponding force const.
> x=loadpdb 1urn_NMRmodified.pdb
Loading PDB file: ./1urn_NMRmodified.pdb
total atoms in file: 1668
> loadoff 8Mureabox.off
Loading library: /Users/ndang/amber11/dat/leap/lib/8Mureabox.off
> solvateoct UREABOX 12.0
solvateOct: Argument #2 is type Double must be of type: [unit]
usage: solvateOct <solute> <solvent> <buffer> [aniso] [closeness]
> solvateoct x UREABOX 12.0
Scaling up box by a factor of 1.284149 to meet diagonal cut criterion
Solute vdw bounding box: 34.528 39.360 47.476
Total bounding box for atom centers: 78.295 78.295 78.295
(box expansion for 'iso' is 33.7%)
Solvent unit box: 46.256 43.053 49.134
Volume: 249294.947 A^3 (oct)
Total mass 105292.482 amu, Density 0.701 g/cc
Added 3697 residues.
> check x
Checking 'x'....
WARNING: The unperturbed charge of the unit: 8.000000 is not zero.
Checking parameters for unit 'x'.
Checking for bond parameters.
Checking for angle parameters.
check: Warnings: 1
Unit is OK.
> addions x Cl- 0
8 Cl- ions required to neutralize.
Adding 8 counter ions to "x" using 1A grid
Grid extends from solute vdw + 5.14 to 11.14
Resolution: 1.00 Angstrom.
grid build: 0 sec
Solvent present: replacing closest with ion
when steric overlaps occur
Calculating grid charges
charges: 2 sec
(Replacing solvent molecule)
Placed Cl- in x at (3.33, 12.53, -15.37).
(Replacing solvent molecule)
Placed Cl- in x at (-14.40, -11.98, 14.05).
(Replacing solvent molecule)
Placed Cl- in x at (10.81, 18.04, -6.41).
(Replacing solvent molecule)
Placed Cl- in x at (-9.05, 12.27, 7.20).
(Replacing solvent molecule)
Placed Cl- in x at (8.12, -1.96, -17.78).
(Replacing solvent molecule)
Placed Cl- in x at (5.94, 13.93, 11.68).
(Replacing solvent molecule)
Placed Cl- in x at (-9.09, 16.54, -7.13).
(No solvent overlap)
Placed Cl- in x at (-19.05, -6.08, -14.45).
Done adding ions.
> alignaxes x
> savepdb x 1urn_8Murea.pdb
Writing pdb file: 1urn_8Murea.pdb
Converting N-terminal residue name to PDB format: NALA -> ALA
Converting C-terminal residue name to PDB format: CVAL -> VAL
> saveAmberparm x 1urn_8Murea.prmtop 1urn_8Murea.inpcrd
Checking Unit.
Building topology.
Building atom parameters.
Building bond parameters.
Building angle parameters.
Building proper torsion parameters.
Building improper torsion parameters.
total 2244 improper torsions applied
Building H-Bond parameters.
Not Marking per-residue atom chain types.
Marking per-residue atom chain types.
(Residues lacking connect0/connect1 -
these don't have chain types marked:
res total affected
CVAL 1
NALA 1
URE 642
WAT 3048
)
(no restraints)
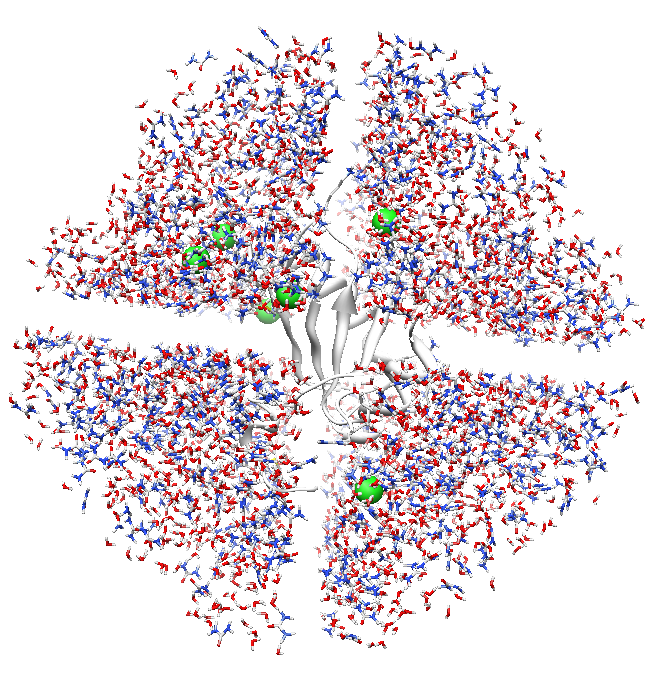
When I check the pdb file in Chimera, there are crossed gaps in the middle
sections of the protein-urea box ( I'm attaching a picture of the system to
this email- but I'm not sure if it will go through the mailling list). Is
that just an artifact? Have anyone encounter this problem before? Should I
go ahead and minimize the system to see if the water and urea molecule
migrate to make up a uniform solution surrounding the protein?
Thanks for your help,
-- Lena Dang' 11
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Picture_2.png)
