Date: Wed, 06 Jul 2011 10:59:32 -0500
Dear Amber users,
I am working on DNA-protein interactions. I have followed the
following steps:
[1] Minimization of water box and counter ions keeping DNA and protein
fixed with a restrained force of 500kcal/mol
[2] Complete minimization of the whole system
[3] Heated the system from 0 to 100 K in 100ps, then from 100 to 200
in 100ps and in the last stage from 200 to 300 in 100ps. So overall
time is 300ps for the heating stage. Some of the simulations I used
1fs time steps, and some of them i used 1fs.
4] Equilibration in 3 steps
First step: Equilibrated the system at constant pressure for
100ps with a restraint force of 10 kcal mol-1 on the protein and DNA.
Second step: Equilibrated the system at constant pressure for
100ps with a restraint force of 1 kcal mol-1 on the protein and DNA.
third step: Free equilibration without any constraints. some of
the simulations I performed 1ns equilibration, some of them I
performed 2ns. Also I used time steps 1 fs and 2fs for some of the
simulations.
[5] production stage simulation for 4ns. Except for one simulation (C)
all other simulations are in NVT ensemble. For prodution stage
simulations I used only 2fs time steps.
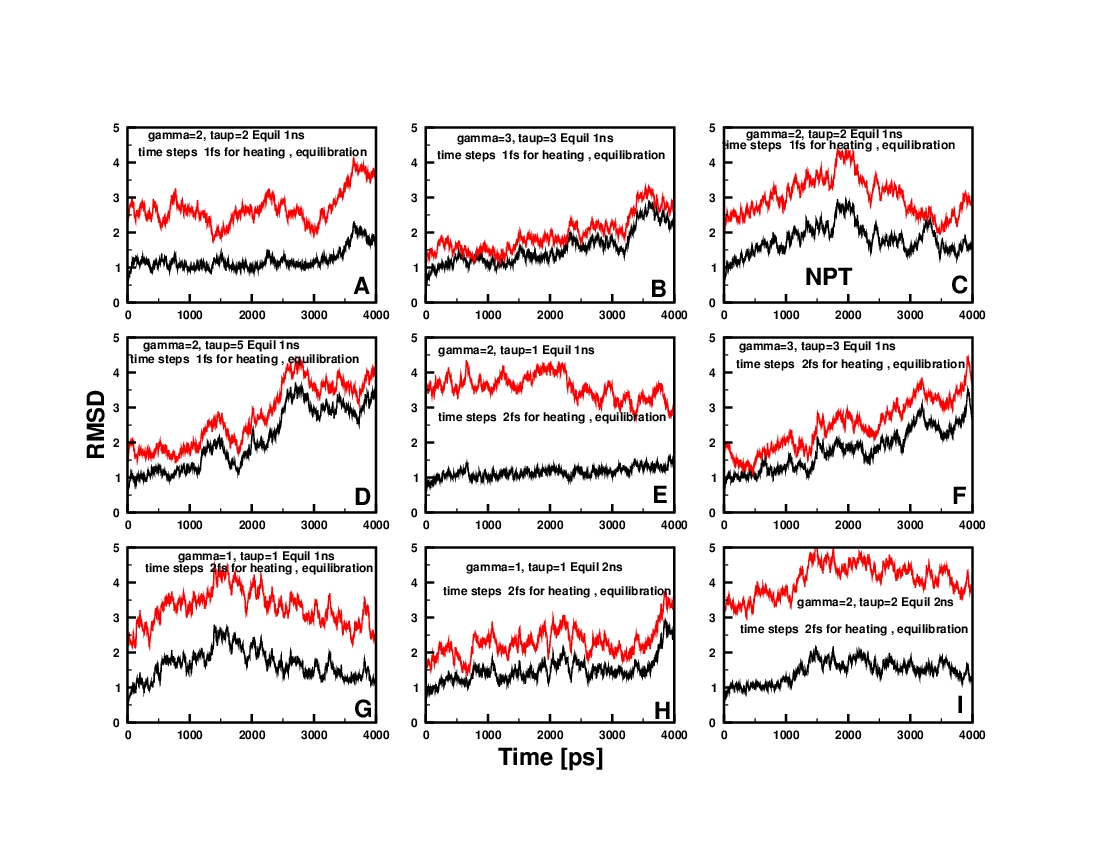
I have performed 9 (A-I) different simulations with different
temperature coupling parameter (gamma_ln ) and pressure couling
parameters (taup).
For simulation A: gamma_ln=2, taup=2, dt=1 fs for heating
and equilbration, Equilibration stage3 1ns
For simulation B: gamma_ln=3, taup=3, dt=1 fs for heating
and equilbration, Equilibration stage3 1ns
For simulation C: gamma_ln=2, taup=2, dt=1 fs for heating
and equilbration, Equilibration stage3 1ns, production
ensemble NPT
For simulation D: gamma_ln=2, taup=5, dt=1 fs for heating
and equilbration, Equilibration stage3 1ns
For simulation E: gamma_ln=2, taup=1, dt=2 fs for heating
and equilbration, Equilibration stage3 1ns
For simulation F: gamma_ln=3, taup=3, dt=2 fs for heating
and equilbration, Equilibration stage3 1ns
For simulation G: gamma_ln=1, taup=1, dt=2 fs for heating
and equilbration, Equilibration stage3 1ns
For simulation H: gamma_ln=1, taup=1, dt=2 fs for heating
and equilbration, Equilibration stage3 2ns
For simulation I: gamma_ln=2, taup=2, dt=2 fs for heating
and equilbration, Equilibration stage3 2ns
Notes: B and F differs only in the time step(dt), G and H differs in
the equlibration stage3 length
I calculated the RMSD deviations withrest to the first frame (BLACK
lines in the RMSD plots) in the production stage and also with repect
to the minimized structure after minimzation step2 (RED lines).. I
have attached the RMSD plots. The RMSD plots shows a large variation
upon changing the simulation parameters..?
My questions:
If these trajectories differ by changing the simulations parameters,
how we can trust these results?
Is there any 'best parameters for gamma_ln and taup.
Is there any problems in the MD protocol which I followed?
What is the reason for the odd behaviour in the RMSD plots and what
should I do to get a stable MD trajectory?
This is the sample inputfile I used for the simulation A:
My inputs: I just changed the respective parameters (gamma, taup, dt,
simulation lenghth) in the input files
Min1:
Minimization solvent + ions Stage 1
&cntrl
imin = 1,
maxcyc = 10000,
ncyc = 5000,
ntb = 1,
ntr = 1,
cut = 12.0
/
Hold the Solute fixed
500.0
RES 1 437
END
END
Min2.in
Minimization whole system
&cntrl
imin = 1,
maxcyc = 10000,
ncyc = 5000,
ntb = 1,
ntr = 0,
cut = 12.0
/
Heat1:
Heating Stage 1
&cntrl
imin = 0,
irest = 0,
ntx = 1,
ntb = 1,
cut = 12.0,
ntr = 1,
ntc = 2,
ntf = 2,
tempi = 0.0,
temp0 = 100.0,
ntt = 3,
gamma_ln = 2.0,
nstlim = 100000, dt = 0.001
ntpr = 1000, ntwx = 1000, ntwr = 1000
/
weak restraints on solute
10.0
RES 1 437
END
END
Heat2:
Heating Stage 2
&cntrl
imin = 0,
irest = 0,
ntx = 1,
ntb = 1,
cut = 12.0,
ntr = 1,
ntc = 2,
ntf = 2,
tempi = 100.0,
temp0 = 200.0,
ntt = 3,
ig = -1,
gamma_ln = 2.0,
nstlim = 100000, dt = 0.001
ntpr = 1000, ntwx = 1000, ntwr = 1000
/
weak restraints on solute
10.0
RES 1 437
END
END
Heating Stage 3
&cntrl
imin = 0,
irest = 0,
ntx = 1,
ntb = 1,
cut = 12.0,
ntr = 1,
ntc = 2,
ntf = 2,
tempi = 200.0,
temp0 = 300.0,
ntt = 3,
ig = -1,
gamma_ln = 2.0,
nstlim = 100000, dt = 0.001
ntpr = 1000, ntwx = 1000, ntwr = 1000
/
weak restraints on solute
10.0
RES 1 437
END
END
Equilibration Stage 1
&cntrl
imin=0,irest=1,ntx=5,
ntc=2,ntf=2,
cut=12.0,
ntb=2, ntp=1, taup=2.0,
ntt=3, gamma_ln=2.0, ig=-1,
temp0=300.0,
tempi=300.0,
ntr=1,
nstlim=100000,dt=0.001,
ntpr=1000, ntwx=1000, ntwr=1000
/
weak restraints on solute
10.0
RES 1 437
END
END
Equilibration Stage 2
&cntrl
imin=0,irest=1,ntx=5,
ntc=2,ntf=2,
cut=12.0,
ntb=2, ntp=1, taup=2.0,
ntt=3, gamma_ln=2.0, ig=-1,
temp0=300.0,
tempi=300.0,
ntr=1,
nstlim=100000,dt=0.001,
ntpr=1000, ntwx=1000, ntwr=1000
/
weak restraints on solute
1.0
RES 1 437
END
END
Equilibration Stage 3
&cntrl
imin=0,irest=1,ntx=5,
ntc=2,ntf=2,
cut=12.0,
ntb=2, ntp=1, taup=2.0,
ntt=3, gamma_ln=2.0, ig=-1,
temp0=300.0,
tempi=300.0,
nstlim=1000000,dt=0.001,
ntpr=1000, ntwx=1000, ntwr=1000
/
MD Production Stage
&cntrl
imin = 0,
irest = 1,
ntx = 5,
ntb = 1,
cut = 12.0,
ntc = 2,
ntf = 2,
temp0 = 300.0,
tempi = 300.0,
ntt = 3,
ig = -1,
gamma_ln = 2.0,
nstlim = 2000000, dt = 0.002
ntpr = 1000, ntwx = 1000, ntwr = 1000
/
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: rmsd1.jpeg)
- application/pdf attachment: RMSD.pdf
