Thanking you
I have downloaded the prep file and frcmod file from the site as you mentioned in your previous mail. The corresponding file name is ATP.prep and frcmod.phos.
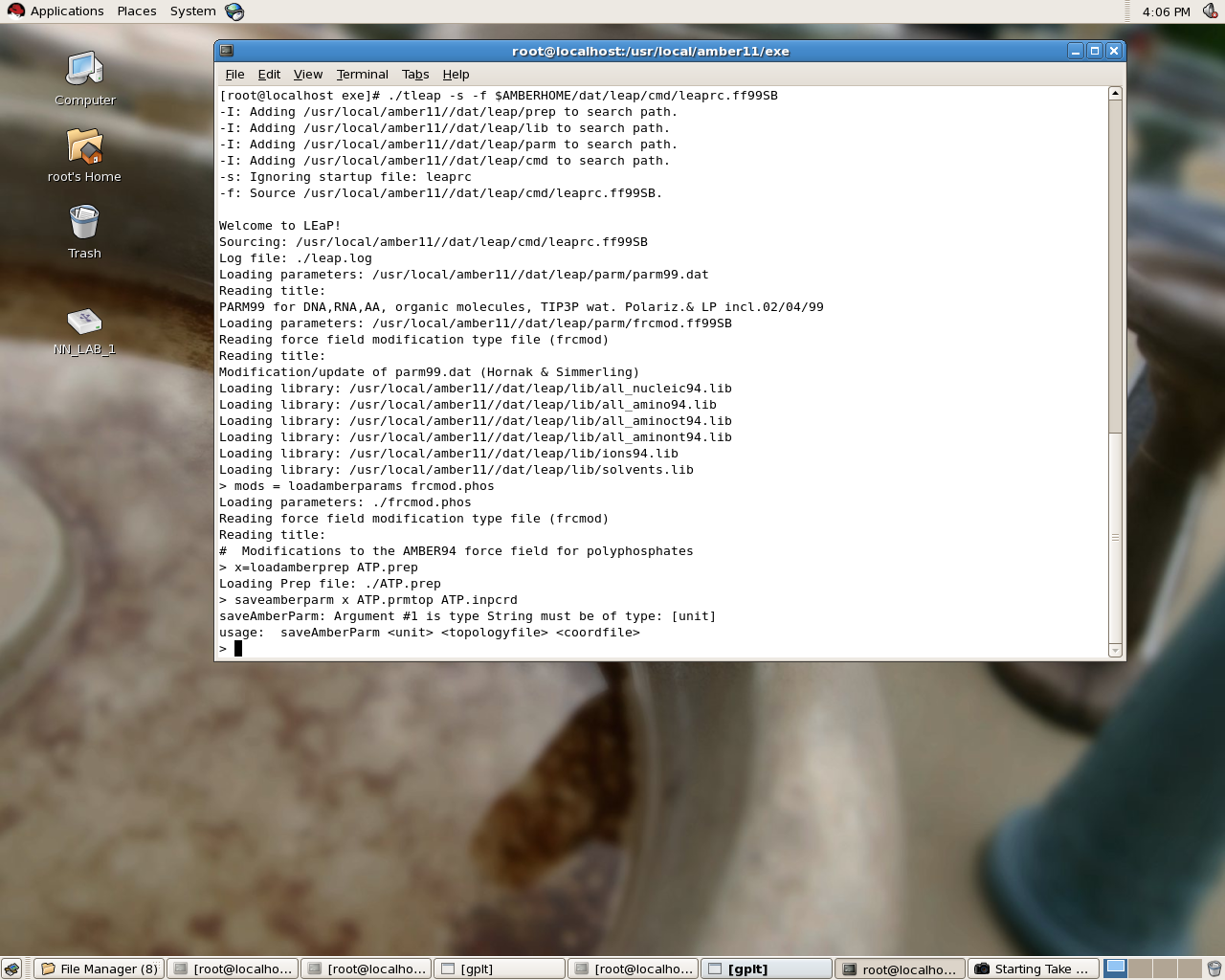
The commands I used to load the prep file and the response I get is as follows:
> mods = loadamberparams frcmod.phos
Loading parameters: ./frcmod.phos
Reading force field modification type file (frcmod)
Reading title:
# Modifications to the AMBER94 force field for polyphosphates
> x=loadamberprep ATP.prep
Loading Prep file: ./ATP.prep
> saveamberparm x ATP.prmtop ATP.inpcrd
saveAmberParm: Argument #1 is type String must be of type: [unit]
usage: saveAmberParm <unit> <topologyfile> <coordfile>
I am also attaching a screenshort. Please find it.
How to solve the problem and save the topology file and coordinate file using the preap file and frcmod file.
With best regards
Sindrila
From: David A. Case <case.biomaps.rutgers.edu>
To: Sindrila Dutta banik <sindrila.duttabanik.yahoo.com>
Sent: Saturday, 11 June 2011 8:09 PM
Subject: Re: Fw: [AMBER] Problem related to the residue template and add ion
On Fri, Jun 10, 2011, Sindrila Dutta banik wrote:
>
> I have downloaded the ".prep" file for ATP from the site as you
> mentioned. I also tried to load ".prep" file to generate the
> corresponding topology file and coordinate file. I used "loadAmberPrep"
> to load the "prep" file as mentioned in AmberTools. But I failed to do
> this. Some how the "loadAmberprep" command is not working properly.
How about some details: what commands did you use? What response did you get
that led you to believe the program was not working correctly? Did you also
load the corresponding frcmod files? Without some real information, it is not
possible to say what went wrong, or why.
...dac
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Mon Jun 13 2011 - 09:30:02 PDT
{kind=link}
