Date: Fri, 25 Feb 2011 11:50:23 +0100
Dear Mark,
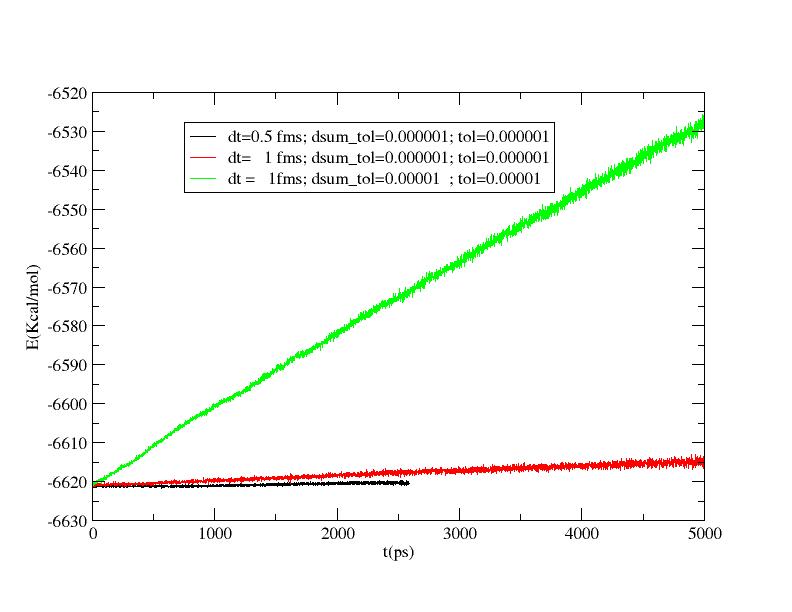
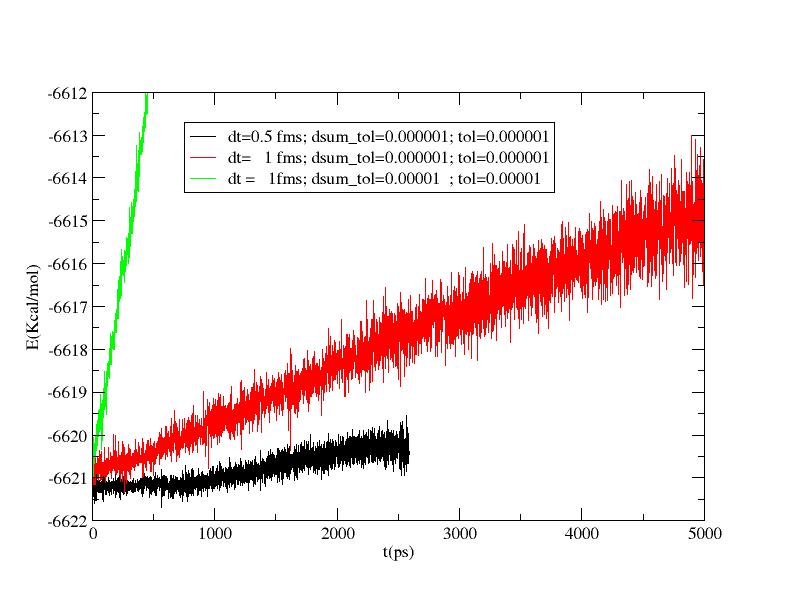
thank you for the link. Fixing dsum_tol =10^-6 and tol=10^-6
there is a sensible improvement of the results.
By the way there is still an energy shift of ~6Kcal/mol in 5 ns with a
time step of 1 fms.
The situation seems to get better reducing the time step (see attachments).
I am using a Car-Parinello scheme (indmeth = 3) for the calculations of
induced dipoles: I am wondering if it is this the problem.
In principle, this method should be faster than the iterative method
(indmeth = 1) but if I have to use a smaller time step, I do not now
know what is more convenient...
Thanks
Ivan
On 02/24/2011 12:37 PM, Mark Williamson wrote:
> Ivan Gladich wrote:
>
>> The strange thing is that the system does not conserve the energy. I am
>> a bit perplex because the energy shift is linear with time (90 Kcal/mol
>> in 5 ns!). The same trend is observable with the smaller time step.
>>
> Try increasing your SHAKE tolerance even further (tol=0.000001) and the
> direct sum tolerance:
>
> &ewald
> dsum_tol=0.000001,
> /
>
> This is quite a brief response, but for more details, have a read of
> this thread:
> http://dev-archive.ambermd.org/200809/0000.html
>
> Regards,
>
> Mark
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
-- ------ Ivan Gladich, Ph.D. Postdoctoral Fellow Academy of Sciences of the Czech Republic Institute of Organic Chemistry and Biochemistry AS CR, v.v.i. Flemingovo nám. 2. 166 10 Praha 6 Czech Republic Tel: +420775504164 e-mail:ivan.gladich.uochb.cas.cz web page:http://www.molecular.cz/~gladich/ -----
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- text/plain attachment: in.md
(image/jpeg attachment: graph_1.jpg)
(image/jpeg attachment: graph_2.jpg)
