Date: Tue, 22 Feb 2011 16:27:30 -0600
Dear , Francois,
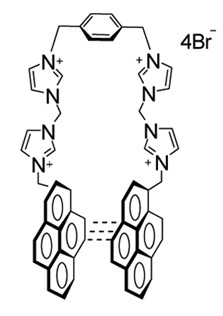
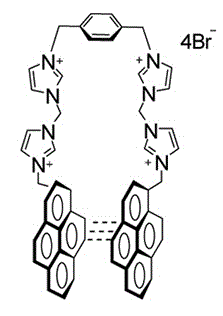
I have attached the molecule in jpg format. The system is a salt whose
charge is neutralized by 4 Br- anions. I have a couple of questions:
[1] Do I need to include the Br- anions in making the paramter file..,
but the exact position of the Br- is unknown...
[2] when I optimize at HF/6-31G*, do i need to include the Br- anion..
Also from literature, Br- aninons helps to maintain the stacked form
of the two pyrene rings,
But when I optimzed, without Br- aninons, the molecules moving away..
Thanks
Rajesh Raju
On Sun, 20 Feb 2011 17:52:57 +0100
FyD <fyd.q4md-forcefieldtools.org> wrote:
> Dear Rajesh Raju,
>
> The problem is not that a program can generate a set of charges or
> not. The problem I wanted to underline is that for such a large &
> multiply charged molecule the conformation obtained after geometry
> optimization is likely to be different to what it is in a solvent or
>
> docked in a protein...
>
> The conformation between multiple cycles is another problem in your
>case...
>
>> Thank you very much for your reply. I have not performed any
>>building
>> block approach for resp charge derivation. I have checked the
>> geomtery. It is a symmetric molecule, with a bridged benzene ring.
>
> Another key problem in this type of 'symmetric' molecule is charge
> equivalencing for chemically equivalent atoms: is it correctly
>applied
> by the program?
>
> You can quickly check chemical equivalencing for your _whole_
>molecule
> using R.E.D. Server:
>
> (i) prepare a PDB file of your structure (with a correct input
> geometry to get a quick result); (ii) upload your PDB input file &
>run
> R.E.D. Server/Ante_R.E.D. 2.0 to generate a P2N file (it will take
>few
> sec); (iii) use the Java applet to display the atom names of the P2N
>
> file.
> Chemical equivalencing is reflected by the first column of atom
>names
> in a P2N file: two chemically equivalent atom should bear a same
>name,
> and after charge derivation they should bear the same charge value
> (when one uses R.E.D.)...
>
>> Can I use RED programme for derving such large molecules..If so can
>>u
>> suggest, the best tutorial for me to follow. I have not used RED
>> before.
>
> R.E.D. & now R.E.D. Server have been developed for RESP & ESP charge
>
> derivation and force field library building. R.E.D. IV & R.E.D.
>Server
> handle quite complex charge derivation by now, and obviously the
> building block approach first defined in the following paper:
> http://www3.interscience.wiley.com/cgi-bin/abstract/109583237/ABSTRACT
>
>> I am have used antechamber quite oftern for small molecules. Is it
>> posssible building block approach for antechamber?
>
> I guess by running antechamber, adapting RESP inputs manually &
> correctly concatenating espot files and re-running antechamber, it
> should be possible...
> The problem here is that one needs to understand the different
>steps.
>
>> My molecules contains 2 pyrene rings, 4 charded imidazole rings, and
>> a briged benzene rings. All the molecules are conncted through
>>-ch2
>> groups. the molecule is symmetrical w.r.t benzene ring. it would be
>> helpful if anyone can suggest a best way to derive charge..
>
> This is difficult to help without a picture of your molecule; Could
>
> you send a drawing for your molecule? However you might look at the
>
> calixarene project in R.E.DD.B. by E. Vanquelef. I am sure you will
>
> find ideas for your own project.
> See http://q4md-forcefieldtools.org/REDDB/projects/F-87/
>
> I hope this helps.
>
> regards, Francois
>
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: receptor.jpg)
(image/gif attachment: receptor.gif)
