Date: Fri, 18 Feb 2011 21:45:49 +0800
Hi all,
Recently I have simulated benzene to estimate its hydration free energy, for the removing charge step, I can create the simulation files and modeling correctly. however, for the second step of the removal vdW, I don’t know how to generate the simulation files. the following is my attempt of removal vdW step.
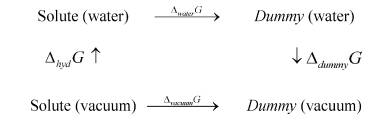
for simulating hydration free energy, on the basis of literatures, one can design thermodynamic cycle as follows:
in terms of above processes, the following equation can be obtained
Now I will display an example of benzene for LJ step.
(1) tleap -f leap.in
(2) cp benw.prm benwv.prm
cp benw.rst benwv.rst
then modify LENNARD_JONES_ACOEF and LENNARD_JONES_BCOEF within benwv.prm file to 0.00.
I use these four files to simulate(not use softcore, i.e. ifsc=0), for lambda=0.1,0.2,0.3,0.4, the simulations work. I inspect the DV/DL variable, the values are very large, up to -2000.
for larger lambda, the simulations fail, the error information reads as follows
Cutoff list exceeds largest sphere in unit cell!!
Big problems with imaging!!
a,b,c = 24.5236717948086 25.2079298792356
21.9999001121865
alpha,beta,gamma = 90.0000000000000 90.0000000000000
90.0000000000000
cutlist,sphere = 11.0000000000000 10.9999500560932
when i modified “solvatebox BNZ SPCBOX 10” to “solvatebox BNZ SPCBOX 12”, the identical error print too.
So, there are two questions
(1) How to create simulation files for the LJ step? in the simulation, whether use softcore or not?
(2) For the non-hydrated benzene, do we also use periodic boundary conditions?
please give your suggestion and your detail operations. thank you very much!
Best regards,
Junjian Miao
School of Chemisty and Chemical Engineering
Nanjing University
Jiangsu province, China
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: clip_image002.jpg)
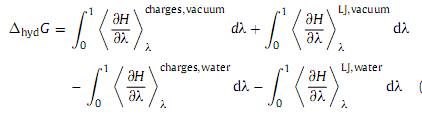
(image/jpeg attachment: clip_image004.jpg)

(image/gif attachment: clip_image005.gif)
