Date: Tue, 15 Feb 2011 14:15:39 +0330
Dear Prof. Roitberg,
Thank you very much for your reply. I am a PhD student of applied mechanics
and intend to study of mechanical properties of DNA under an external force.
Because of our limited computer resource, I decided to start my simulation
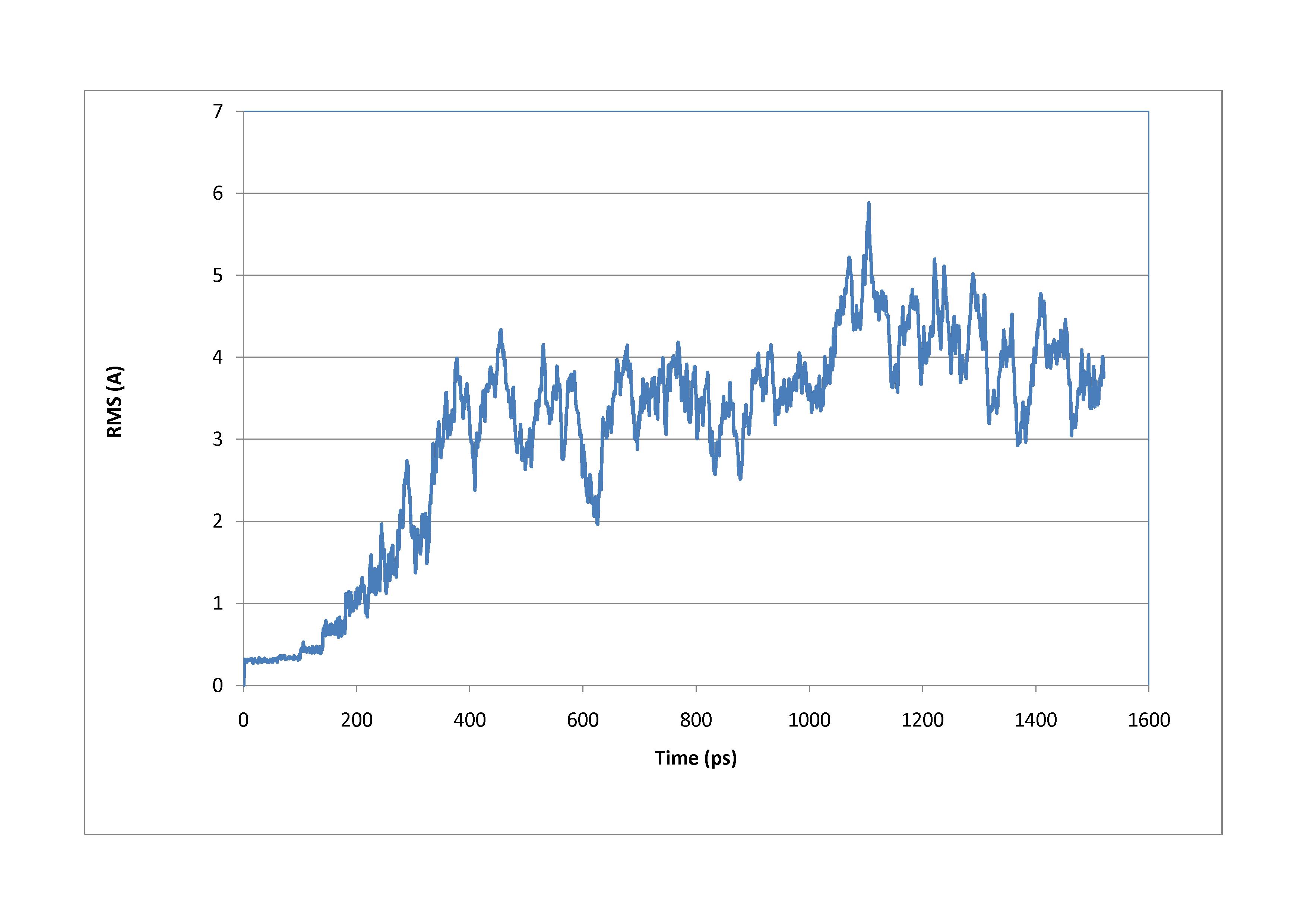
in implicit environment. I tried to equilibrate an (ACTG)3 dodecamer for
about 1.5 ns, before stretching it. I attach the obtained plot for root mean
square deviation of DNA backbone vs. time.
Also, please point that H-bond analyses by VMD show that at the end of the
equilibrium, number of hydrogen bonds between complementary nucleotides is
less than 30 (about 24) for the (ACTG)3, in spite of using cutoff
distance=3.5 Å & angle cutoff=25 degree.
Your advices in this regard is highly appreciated.
On Tue, Feb 15, 2011 at 11:53 AM, Adrian Roitberg <roitberg.qtp.ufl.edu>wrote:
>
>
> On 2/15/11 9:15 AM, Ali M. Naserian-Nik wrote:
> > Hi all,
> >
> >
> > I am going to pull double stranded DNA in implicit environment, using
> > generalized Born/surface area model. Now I am not sure about two issues:
> Dear Ali
>
> While you are of course free to do this, I strongly recommend against
> it. DNA/RNA are not quite stable under current GB implementations, so a
> lot of what you will be seeing are effects of the initial
> structure/dynamics. Maybe try to run a long MD for the unstretched DNA
> first and see if it behaves 'properly' before going too far. If this
> test does not give you stable enough trajectories, think about going
> into explicit solvent runs.
>
> >
> > 1- I think that by this means, the hydrophobic contribution to solvation
> > free energy is taken to account for my GB calculations. Does the
> > basestacking effect for DNA is accounted in the force-extension curves,
> > which is obtained from steered MD simulations?
> In short, yes. See the list for a similar question before regarding
> 'stacking' in DNA. IN the classical force fields this stacking is not
> explicitly treated as such, but rather as a set of vdw and
> electrostatics interactions.
>
>
> >
> > 2- Can I employ the ff99bsc0 force field, or I should use ff99SB force
> field
> > as suggested by the users’ manual?
> ff99SB is just for proteins, not for nucleic acids. try using ff99bsc0
> >
> >
> > I'd be so grateful if anyone could help me on these issues.
>
> Let me know if we can be of more help.
> >
> > Kind regards,
> >
> > Ali
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
>
> --
> Dr. Adrian E. Roitberg
> Associate Professor
> Quantum Theory Project, Department of Chemistry
> University of Florida
>
> on Sabbatical in Barcelona until August 2011.
> Email roitberg.ufl.edu
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- Ali
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: RMS_-_actg_3_gb1_equil.jpg)
