Date: Fri, 07 Jan 2011 19:32:58 +0100
Hi everybody,
I'm sure that some of you must have made a similar observation.
Following equilibration, and during the production run the backbone rmsd of the protein is drifting. I'm attaching two snapshots for easy reference. The equilibration run was over 6ns and the production run over 14ns. The md.in files are identical.
Any ideas regarding this behaviour?
Many thanks
George
EQUILIBRATION: md.in
heat 3ogn-3og
&cntrl
imin=0,irest=1,ntx=5,
nstlim=1000000,dt=0.002,
ntc=2,ntf=2,
cut=8.0, ntb=2, ntp=1, taup=2.0,
ntpr=1000, ntwx=1000,
ntt=3, gamma_ln=2.0, ig=-1,
temp0=300.0,
/
================
PRODUCTION: md.in
heat 3ogn-3og
&cntrl
imin=0,irest=1,ntx=5,
nstlim=1000000,dt=0.002,
ntc=2,ntf=2,
cut=8.0, ntb=2, ntp=1, taup=2.0,
ntpr=5000, ntwx=5000,
ntt=3, gamma_ln=2.0, ig=-1,
temp0=300.0,
/
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
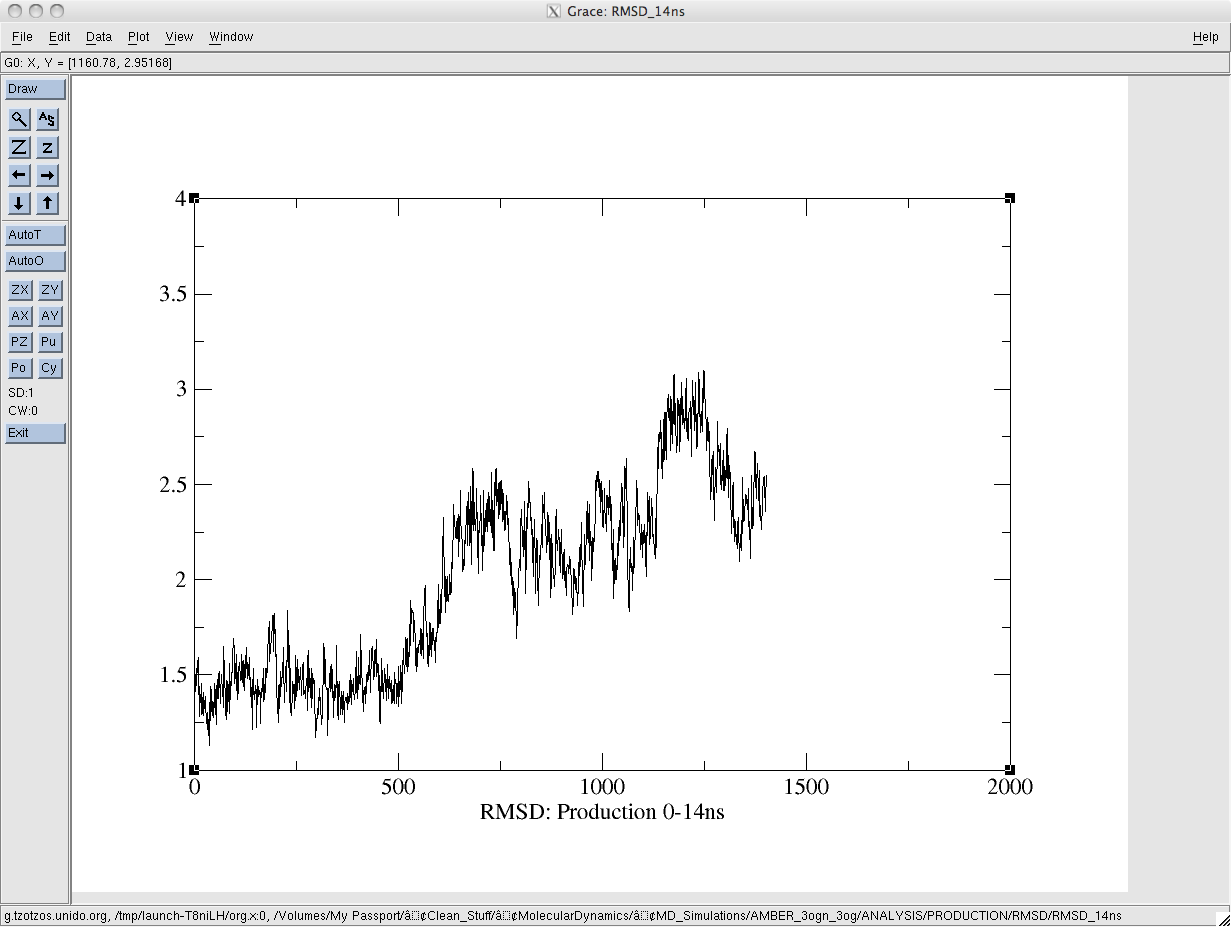
(image/png attachment: prod_rmsd.png)
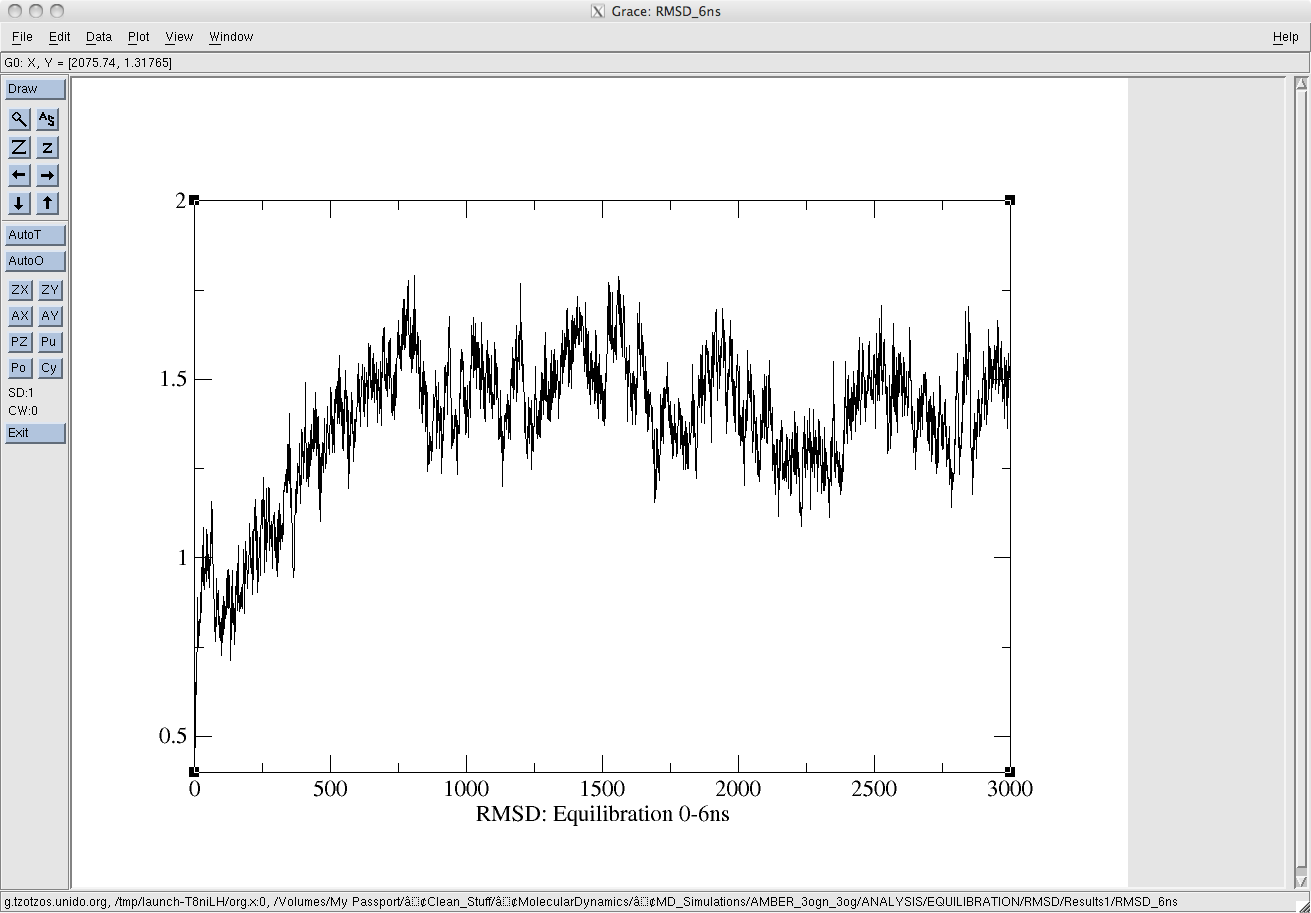
(image/png attachment: equil_rmsd.png)
