Date: Mon, 29 Nov 2010 20:19:16 +0100
Dear Dr. Simmerling,
The replicas have the same input coordinate file, namely the restart
file from the NPT run I used for relaxing the system. So there is no
way the box sizes could be different.
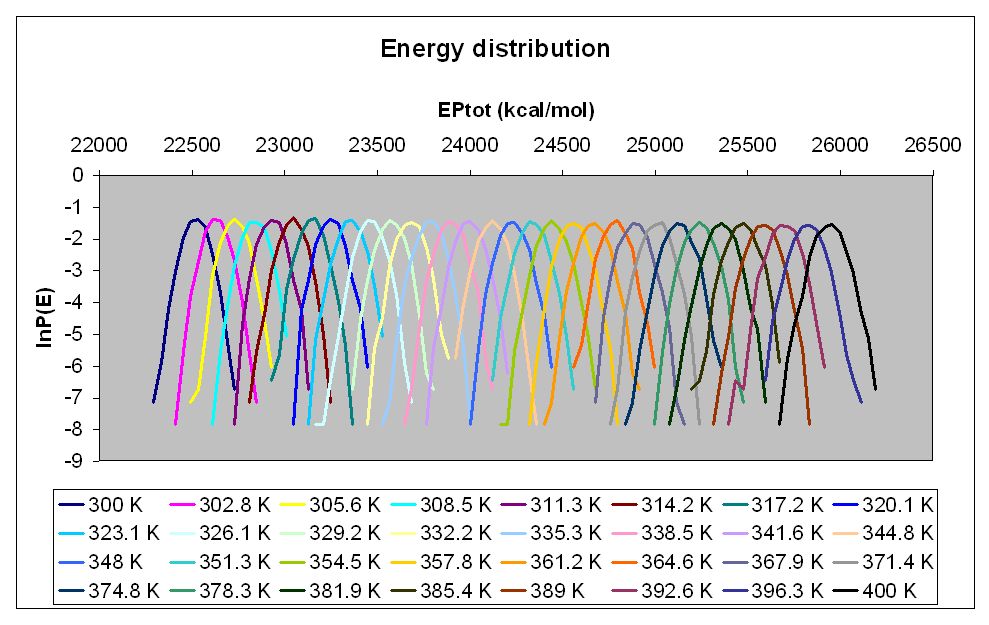
Following your advice, I've run a 5 ns md simulation at each
temperature, and all of the simulations finished correctly. I have
created the energy distribution histogram of each run as you
suggested, and there is sufficient overlap between the potential
energies (as far as I can tell). I have enclosed an image of the
histograms.
Since the md runs never crashed, I think the problem would be
something regarding the replica exchange step.
Any advice what should be the next thing I look into?
Thank you in forward,
Gabor Janzso
Quoting "Carlos Simmerling" <carlos.simmerling.gmail.com>:
> it's still unclear to me if the initial structures have different volumes or
> not- if yes, this can make exchanges very difficult.
>
>
> I suggest running the identical simulation without remd- meaning set up all
> of the repliacs and temepratures, but do not use remd. check to make sure it
> is still stable (and verify that REMD is the problem). from this, extract
> potential energies from the output files and histogram all of them to ensure
> that there is overlap between neighbors.
>
>
> On Tue, Nov 23, 2010 at 1:07 PM, Janzsó Gábor <janzso.brc.hu> wrote:
>
>> Dear Mr. Simmerling,
>>
>> I am sorry if I wasn't clear, my goal is to run an NVT study. The NPT
>> part was only to relax the system after solvating the peptide in the
>> TFE, just as the tutorials and the manual suggest.
>>
>> Regarding your second advice, I am not sure how to create the
>> histogram of the potential energies if the replicas do not behave as
>> expected? Should I run simple md runs at each temperature instead? How
>> long such a run shoul be?
>>
>> I am also almost sure that the phase transition is not the cause of my
>> problem, since I also tried to run my simulation between 300K and 350K
>> (with 32 replicas), and 350K is just below the boiling point of TFE.
>> My first guess was the replicas were too far away from each other, and
>> because I have only limited computational capacity at my disposal, my
>> only option was for sampling the temperatures more frequently,
>> decreasing the temperature range. Regardless, on lower temperatures,
>> with smaller deltaT values, the same behavior was observed.
>>
>> best regards,
>>
>> Gabor Janzso
>>
>>
>> Quoting "Carlos Simmerling" <carlos.simmerling.gmail.com>:
>>
>> > it's very important to study REMD examples in the literature before
>> trying
>> > something very complex like what you want. First, most studies are done
>> at
>> > NVT. Check work by Angel Garcia if you want to include pressure effects.
>> > Second, it is important to carefully histogram your potential energies
>> for
>> > the replicas. Like you are trying to sample across a phase transition,
>> which
>> > is quite challenging. Almost certainly this was not included in your
>> method
>> > for selecting the replica temperatures (which you have not told us
>> about).
>> >
>> > perhaps there is something else going on- but I think the first step is
>> to
>> > try NVT.
>> >
>> > 2010/11/23 Janzsó Gábor <janzso.brc.hu>
>> >
>> >> Dear Amber Users!
>> >>
>> >> I run into a problem with Amber REMD. I am using Amber 9, and I do not
>> >> have the option to upgrade to 11, so any solution working on Amber 9
>> >> would be much appreciated.
>> >> So, I try to run an NVT simulation of amyloid beta 1-42 (Ab1-42) in
>> >> explicit TFE solvent.
>> >>
>> >> I downloaded the mol2 file I found on REDDB (project code W-16), I
>> >> used packmol to put 256 molecule into a=30.125 cubic box, and then
>> >> relaxed the box at 300 K. (first heated up with NVT, than relaxed with
>> >> NPT) I saved the output as a lib file, than used it as the solvent box
>> >> to solve the peptide. I've run some NVT and NPT dynamics to see if its
>> >> stable, and it was, at least up to 400K. At 450K or 500K the
>> >> simulation stopped, the output said SANDER BOMB stopped the run or
>> >> something like that. I figured it might be ok, because the boiling
>> >> point of TFE is at 78°C, and the studies I have found used the
>> >> temperature range of 300K-400K for TFE solvent simulation.
>> >>
>> >> So, I set up a REMD using 32 replicas between 300K and 400K, with
>> >> Berendsens thermostat (1 ps coupling) SHAKE is on, exchange attempts
>> >> at every 2 ps, and chirality restraints and trans-omega restraints are
>> >> applied.
>> >> The simulation starts normally, but around the first ten-twenty
>> >> exchange attempts some replicas heat up like insane. The REMD keeps on
>> >> running, but three replicas are at ~600 000K (!) - and obviously they
>> >> don't participate in the exchanges anymore, so the simulation does not
>> >> stop.
>> >> The curious thing is, that it always happens after a successful
>> >> exchange, and it happens always to the same replicas. What I mean, in
>> >> the rem.log file where all the replicas and the relevant info is
>> >> listed, the 9th, 17th and 25th replicas heat up. Always this three. I
>> >> tried it with different parameters, for example the timestep was
>> >> reduced to 1 ps, the iwrap option was turned off, the vlimit was
>> >> reduced to 10, but nothing helped, the same replicas systematically
>> >> has gone wild every time.
>> >>
>> >> If anyone has any idea, what could be the reason for this phenomenon,
>> >> it would be much appreciated.
>> >>
>> >> Thanks in advance
>> >>
>> >> Gabor P. Janzso
>> >> PhD student
>> >> Institute of Biophysics,
>> >> Biological Research Center
>> >> H-6726, Szeged, Temesvári krt. 62.
>> >>
>> >> Janzsó Gábor Péter
>> >> PhD hallgató
>> >> Szegedi Biológiai Központ,
>> >> Biofizikai Intézet
>> >> 6726, Szeged, Temesvári krt. 62.
>> >>
>> >> ----------------------------------------------------------------
>> >> This message was sent using IMP, the Internet Messaging Program.
>> >>
>> >>
>> >> _______________________________________________
>> >> AMBER mailing list
>> >> AMBER.ambermd.org
>> >> http://lists.ambermd.org/mailman/listinfo/amber
>> >>
>> > _______________________________________________
>> > AMBER mailing list
>> > AMBER.ambermd.org
>> > http://lists.ambermd.org/mailman/listinfo/amber
>> >
>>
>>
>>
>> Gabor P. Janzso
>> PhD student
>> Institute of Biophysics,
>> Biological Research Centre
>> Hungarian Academy of Sciences Szeged
>> H-6726, Szeged, Temesvári krt. 62.
>>
>> Janzsó Gábor Péter
>> PhD hallgató
>> Szegedi Biológiai Központ,
>> Biofizikai Intézet
>> 6726, Szeged, Temesvári krt. 62.
>>
>> ----------------------------------------------------------------
>> This message was sent using IMP, the Internet Messaging Program.
>>
>>
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
Gabor P. Janzso
PhD student
Institute of Biophysics,
Biological Research Centre
Hungarian Academy of Sciences Szeged
H-6726, Szeged, Temesvári krt. 62.
Janzsó Gábor Péter
PhD hallgató
Szegedi Biológiai Központ,
Biofizikai Intézet
6726, Szeged, Temesvári krt. 62.
----------------------------------------------------------------
This message was sent using IMP, the Internet Messaging Program.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: Energy_distribution.jpg)
