Date: Thu, 21 Oct 2010 16:09:14 -0700
Hello Ross,
It's really strange that I guess I solved the problem.
I started with the two-carbon unit cell.
1. I made prepi file from the two-carbon
================================
0 0 2
This is a remark line
molecule.res
GRA INT 0
CORRECT OMIT DU BEG
0.0000
1 DUMM DU M 0 -1 -2 0.000 .0 .0 .00000
2 DUMM DU M 1 0 -1 1.449 .0 .0 .00000
3 DUMM DU M 2 1 0 1.522 111.1 .0 .00000
4 C c1 M 3 2 1 1.540 111.208 180.000 0.000000
5 C1 c1 M 4 3 2 1.421 90.000 -178.629 0.000000
LOOP
IMPROPER
DONE
STOP
=========================================================
2. I generate a graphite structure with
PropPDB -p 2c.pdb -o 20.pdb -X 2.461 -Y 2.461 -Z 6.708 -a 90 -b 90 -g
120 -ix 20 -iy 20 -iz 1
At this point, I just tried to make a bigger one about the
edge-to-edge distance >50A.
3. Generate prmtop and inpcrd file by
---------------------------------------------------
source leaprc.ff99SB
source leaprc.gaff
loadamberprep c2.prepi
loadamberparams c2.frcmod
gr=loadpdb 20.pdb
bondByDistance gr 1.5
savepdb gr 20amb.pdb
solvatebox gr TIP3PBOX {0 0 20}
saveamberparm gr c20.prmtop c20.inpcrd
--------------------------------------------------
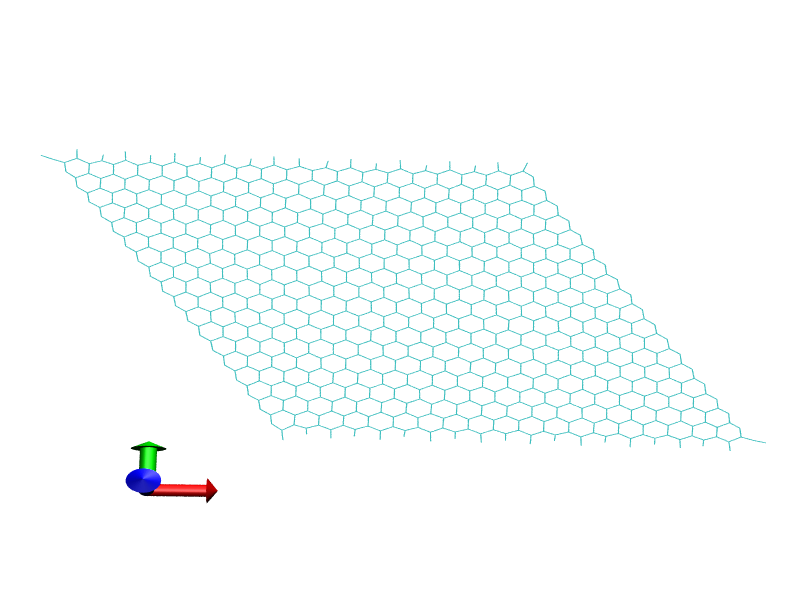
And I can see no weird bonds between upper edges and lower edges.
4. I used smaller restraint about 10 and it passed all minimization
and heating stages without breaking the graphite structure. You can
see this picture. This is about 50ps after the heating.
In sum up, larger structure can avoid making wrong bonds in leap.
I guess there is threshold value to make a bond in the inside of leap.
Thank you so much.
Bongkeun Kim
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: c20.png)
