Date: Wed, 22 Sep 2010 10:44:32 +0800
Thank you very much for your help! In my case, the water box is a truncated
octahedron.
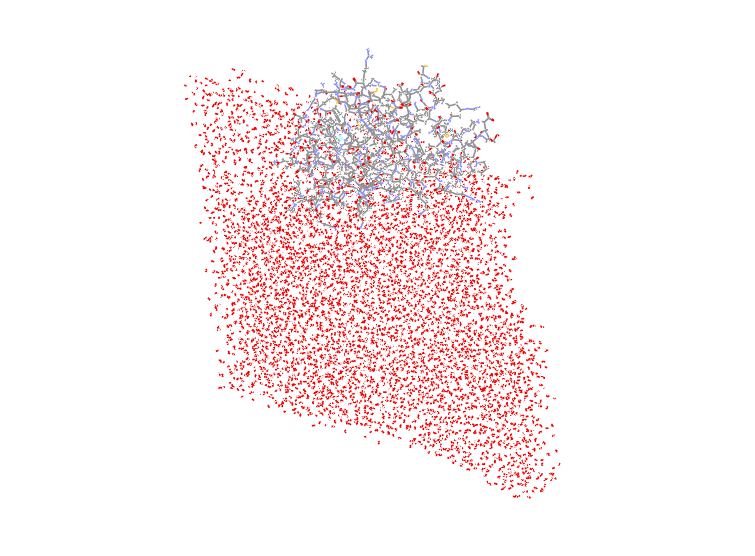
I have also tried using the "image center" instead of "image center
familiar", and the water box turns out to be a rhombohedron with the protein
at the edge. I have attach the picture of one of the pdb files. It's
amazing. Shouldn't the protein be in the center of box when both "center"
and "image center" are using?
Is there any kind of cases in which water molecule can distribute
homogeneous?
Looking forward to your reply!
Best Regards
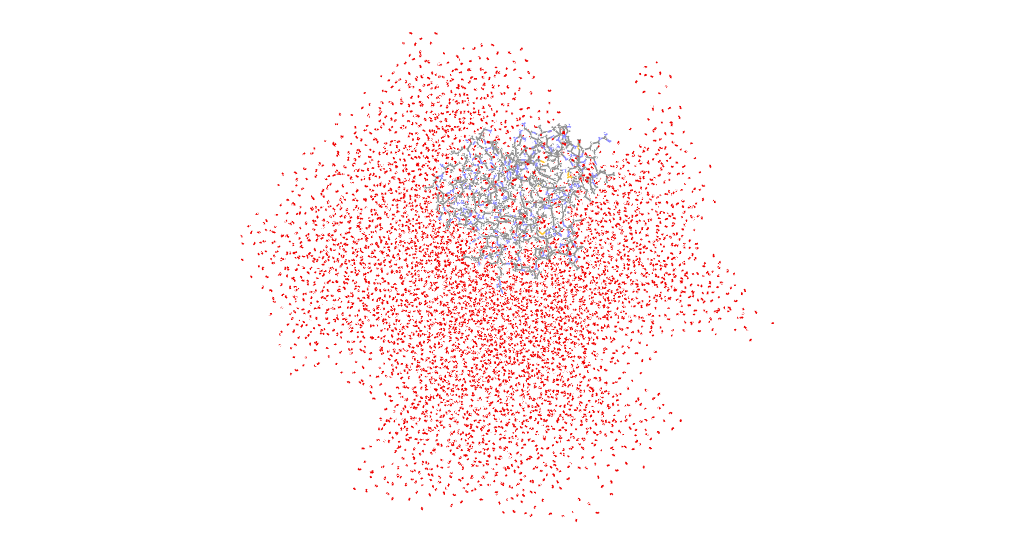
-- Zhang Bingbing On Tue, Sep 21, 2010 at 10:42 PM, Daniel Roe <daniel.r.roe.gmail.com> wrote: > It looks like your system is using a rectangular box. If this is the > case try using "image center" instead of "image center familiar". I'm > not 100% sure but I think the 'familiar' keyword is only for truncated > octahedral boxes. > > -Dan > > On Sun, Sep 19, 2010 at 10:57 AM, bingbing zhang > <zbingbing.ruc.gmail.com> wrote: > > Dear, > > > > I am using AMBER9 to equilibrate and run production dynamics on a > protein. > > Everything seems normal during the equilibration progress, but the PDB > files > > generated by "trajout" have some problems. I run the MD simulation with > > "iwrap=1". > > > > Form figure1, the picture of one of these PDB files, it is obvious to > > find that the protein is at the edge of the box and some atoms stay > outside > > the water TIP3P box. It seems like there is a big ball-like thing in the > > space. > > > > So I try adding " center :1-150" and " image center familiar" to the > > ptraj file to locate the solute molecule into box. Then I get the figure2 > > and 3, picture of new pdb file. In the new pdb, the water molecules are > not > > homogeneous. I want to get two or more box show in pdb, but I don't know > how > > to get it. > > > > Does someone have any suggestion to overcome the problems? > > > > Looking forward to getting your help! > > > > Thank you very much in advance! > > > > > > Best Regards > > > > Zhang Bingbing > > > > _______________________________________________ > > AMBER mailing list > > AMBER.ambermd.org > > http://lists.ambermd.org/mailman/listinfo/amber > > > > > > _______________________________________________ > AMBER mailing list > AMBER.ambermd.org > http://lists.ambermd.org/mailman/listinfo/amber >
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: center-nof.png)
(image/png attachment: center-nof-2.png)
