Date: Sat, 31 Jul 2010 17:03:39 -0700 (PDT)
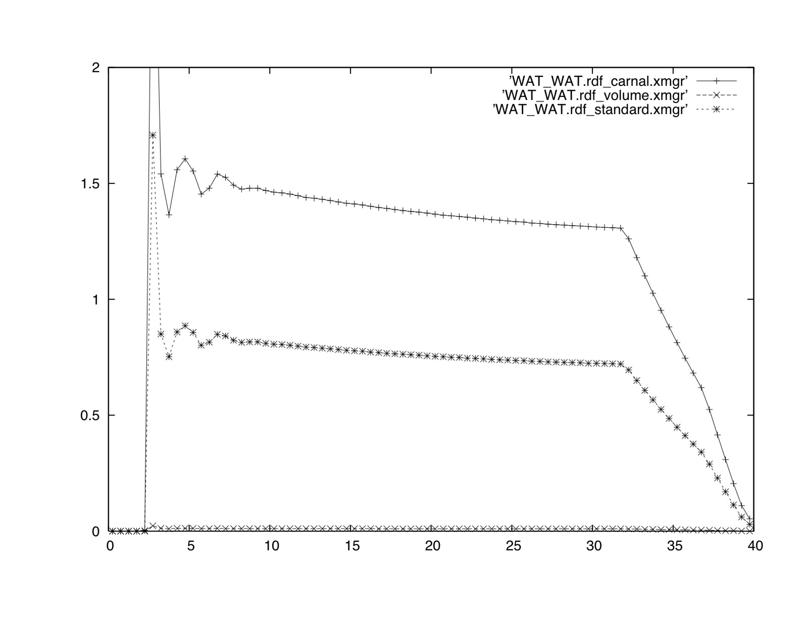
Hi Thanks a lot for the previous comments. My molecule has indeed a radius of almost 30 Å. I made 2 rdf calculations ONE to track water inside the macromolecule :WAT.O :1.H19, the second to see the RDF of the solute wrt to cdm i.e :1-121 :1.H19. Both give almost zero values. There is a paper that gives the radial density distribution of the same molecule using the center of mass as the reference and gives values decreasing from 1200 to 0 kg/m**3. Which output I should compare to this paper? (standard, volume or carnal). I really do not know what is "backward compatibility" I do not know how the RDF of :WAT.O :WAT.O should look like, which file i should see, (carnal,standard or volume)? Enclosed please find a figure of :WAT.O :WAT.O, from my md calculation, please could you comment/ give advice on this figure and the unclear previous points? Many thanks Regards ________________________________ From: Thomas Cheatham <tec3.utah.edu> To: AMBER Mailing List <amber.ambermd.org> Sent: Fri, July 30, 2010 2:44:56 AM Subject: Re: [AMBER] RDF 3 files and 3 columns > all volume and standard files give me RDF=0, only carnal has got some non zero Perhaps your solute has a radius > 20A and there is no water inside? As a first check, do the RDF of the water only. If this doesn't look "normal" something funny is happending. If this looks good then do the RDF to a surface residue... Try to experiment; also drop snapshots for visualization to make sure things look OK. Note that if there is only a few waters inside the solute, this will show as values close to zero. As your radius expands, the ratio to bulk decreases rapidly! _______________________________________________ AMBER mailing list AMBER.ambermd.org http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: WAT.O_WAT.O.jpg)
