Date: Mon, 19 Jul 2010 14:04:23 +0200 (CEST)
Dear Amber Users and Developers,
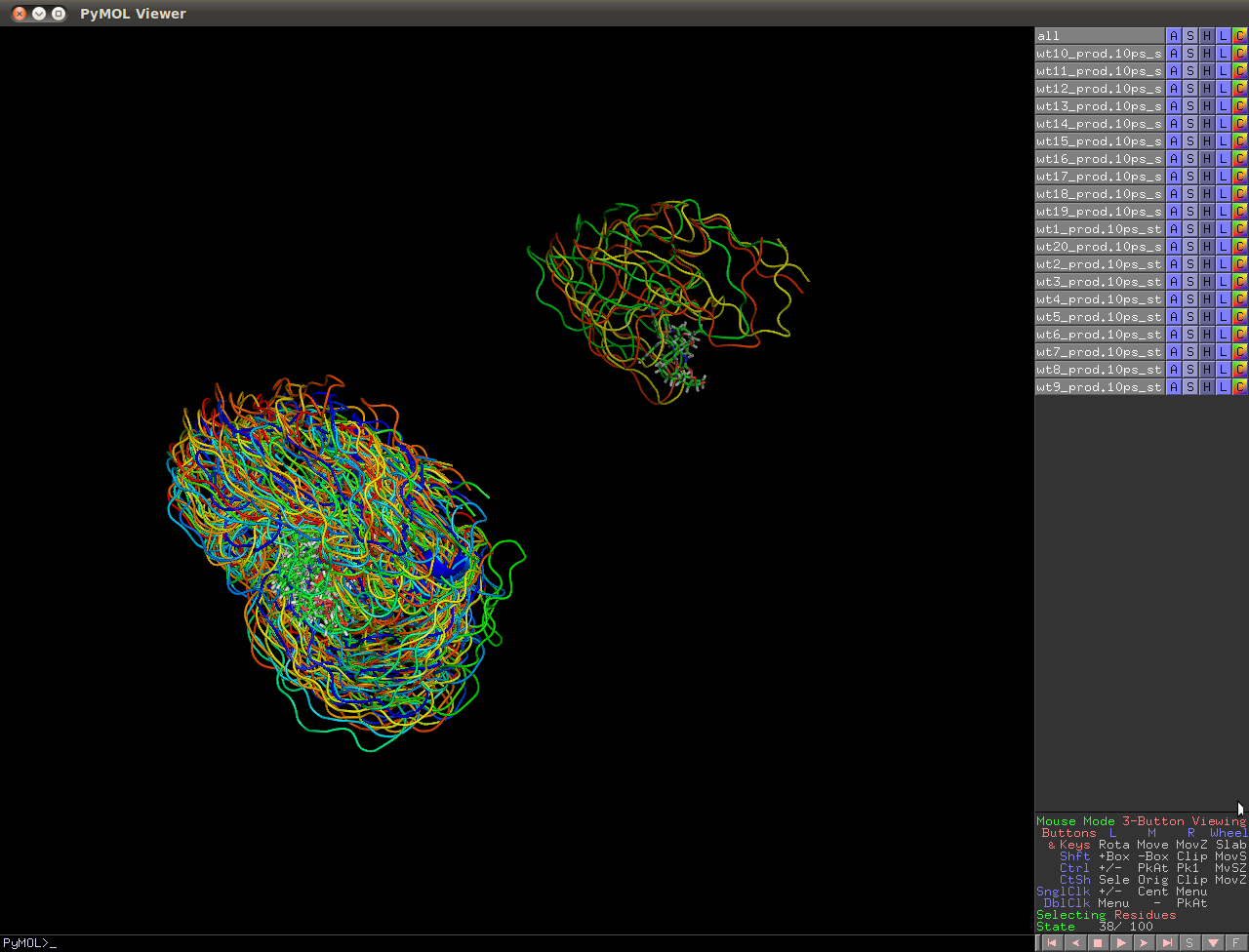
I'm still having trouble with a well know issue: A dimeric protein diffuses across the periodic boundary and is split into monomers (as can be seen in the attached png).
The trajectory is needed for a MMPBSA.py calculation.
I know several possibilities to circumvent this issue in principle but none works.
1. The center-of-mass motion should be removed by default so that the protein stays in the box center - please correct me if I'm wrong.
(Or do I have to remove comm more often than default?)
2. iwrap=1 should map all atoms into the primary box when writing the trajectory.
3. tleap can be used to map the atoms back into the primary box - this is somewhat tricky (by centering two times - on time without and then with ligand) implemented in the MMPBSA.py script:
trajin production.mdcrd 1 1000 1
strip :WAT:Cl-:CIO:Cs+:IB:K+:Li+:MG2:Na+:Rb+
center :1-198 mass origin
image origin center
center :1-199 mass origin
image origin center
rms first mass :1-198
average _MMPBSA_avgcomplex.pdb pdb
trajout _MMPBSA_complex.mdcrd nobox
In the end, I still run into problems with my energy calculations (as can be seen in the attached pdf - 20 simulations with 3 of them having PBC effects).
Can anybody tell me how to prepare the trajectories so that monomer1, monomer2 and ligand coordinates are in one box?
Thanks for any help
Oliver
___________________________________________________________
Neu: GMX De-Mail - Einfach wie E-Mail, sicher wie ein Brief!
Jetzt De-Mail-Adresse reservieren: http://portal.gmx.net/de/go/demail
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- application/pdf attachment: h_cum_f.pdf
(image/png attachment: pbc_effect.png)
