Date: Tue, 30 Mar 2010 13:11:17 +0800 (CST)
Hi case,
Thanks for your reply.
AMBER version I use is AMBER FF03 force field,and sander is the programe to run MD simulation on villin headpiece, the protein I'm interested in.
I first minimized my system,and heated it to 300K in 20 ps ,then equilibrated it with NPT for 5ns.Finally, performed production run with NVT and it stopped at 4ns without "iwrap=1".
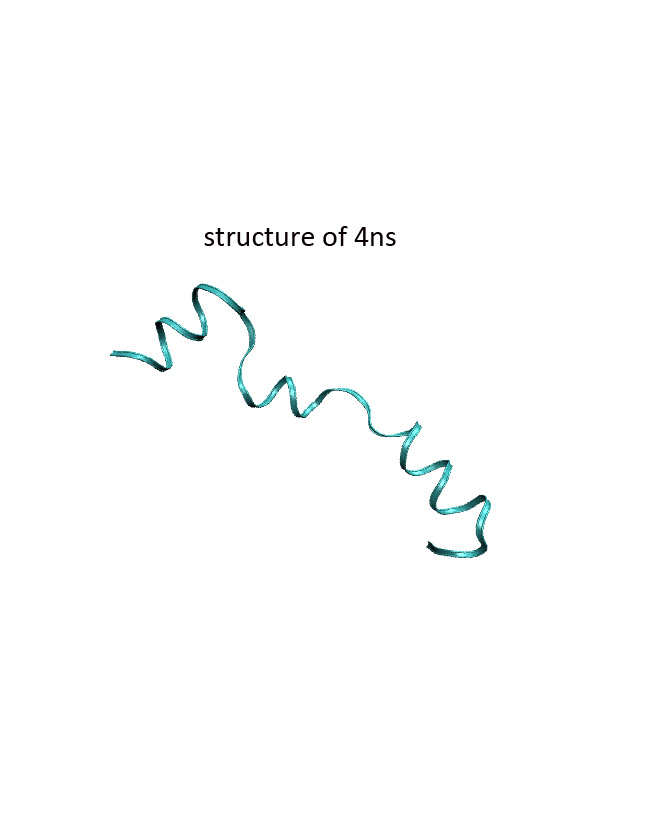
The attachment is two pictures, one for XRD structure of villin headpiece, the other for simulated structure at 4ns production run. Output file was recovered due to my careless, so sorry for that.
Thanks in advance.
Sincerely,
Jianing
在2010-03-29 22:19:26,case <case.biomaps.rutgers.edu> 写道:
>On Mon, Mar 29, 2010, Nancy wrote:
>>
>> One was without iwrap=1 and the system stopped at 4ns with error info:
>> coordinates of the atoms are too large,
>> so MD exited.
>
>Please copy and paste the exact error message; also let us know what program
>you are running, and with which version of Amber.
>
>Ben's reply is basically correct, but if you are getting the error he refers
>to after only 4ns, that would be rather unusual.
>
>...thanks...dac
>
>
>_______________________________________________
>AMBER mailing list
>AMBER.ambermd.org
>http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: 4ns.jpg)

(image/jpeg attachment: xrd_villin_.jpg)
