Date: Tue, 9 Feb 2010 13:02:48 -0800 (PST)
Hi all,
I am new to Amber and have just been trying out Thomas Steinbrechers Thermodynamic Integration tutorial on the transformation of benzene to phenol. I used the instructions to measure the energy difference between hexen-1-ol and heptan-1-ol binding to the mouse major urinary protein (MUP). The free energy values I got agreed with the experimental values obtained via ITC. However, the RMS error seems quite large in comparison. Thus, I wanted to know if there was a way to reduce this value, or whether it was unavoidable statistical noise.
I have calculated the RMS by subtracting the RMS of the ligand in water from that of the complexed ligand. This was then fed into the script on the last page of the tutorial. Is this the correct way to calculate the RMS? Would better error values be obtained by increasing the number of lambda points sampled, or by increasing the equilibration time?
My input files for the complex in step2 are as follows:
density minlibration
&cntrl
imin = 1, ntx = 1, ntmin=2,
maxcyc=500,
ntpr = 1,
ntf = 1, ntc = 1,
ntb = 1, cut = 9.0,
icfe=1, clambda = 0.8,
ifsc=1,
crgmask=':158.H14',
scmask=':158.H14',
&end
-------------------------------------------------
density equilibration
&cntrl
imin = 0, ntx = 1, irest = 0, ntmin=2,
ntpr = 2500, ntwr = 10000, ntwx = 0,
ntf = 1, ntc = 1,
ntb = 2, cut = 9.0,
nstlim = 25000, dt = 0.001,
temp0 = 300.0, ntt = 3, gamma_ln = 5,
ntp = 1, pres0 = 1.0, taup = 0.2,
icfe=1, clambda = 0.1,
ifsc=1,
crgmask=':158.CT7,H15,H16,H7',
scmask=':158.CT7,H15,H16,H7',
&end
-------------------------------------------------
NPT production
&cntrl
imin = 0, ntx = 5, irest = 1, ntmin=2,
ntpr = 10000, ntwr = 100000, ntwx = 10000,
ntf = 1, ntc = 1,
ntb = 2, cut = 9.0,
nstlim = 100000, dt = 0.001,
temp0 = 300.0, ntt = 3, gamma_ln = 2,
ntp = 1, pres0 = 1.0, taup = 2.0,
icfe=1, clambda = 0.1,
ifsc=1,
crgmask=':158.H14',
scmask=':158.H14',
&end
-----------------------------------------------------
My final results are;
l: 0.00 (w=0.050) cont: 0.0050+--0.0025
l: 0.10 (w=0.100) cont: 0.0020+--0.0050
l: 0.20 (w=0.100) cont: -0.0060+--0.0070
l: 0.30 (w=0.100) cont: 0.0000+--0.0070
l: 0.40 (w=0.100) cont: 0.0020+--0.0080
l: 0.50 (w=0.100) cont: -0.0030+--0.0080
l: 0.60 (w=0.100) cont: -0.0040+--0.0080
l: 0.70 (w=0.100) cont: -0.0020+--0.0090
l: 0.80 (w=0.100) cont: -0.0060+--0.0080
l: 0.90 (w=0.100) cont: -0.0010+--0.0060
l: 1.00 (w=0.050) cont: 0.0020+--0.0030
Total DV/DL:-0.011+--0.0715
Step2
l: 0.00 (w=0.050) cont: 0.0055+--0.0075
l: 0.10 (w=0.100) cont: -0.0810+--0.0150
l: 0.20 (w=0.100) cont: -0.1730+--0.0270
l: 0.30 (w=0.100) cont: -0.1860+--0.0490
l: 0.40 (w=0.100) cont: -0.1150+--0.0900
l: 0.50 (w=0.100) cont: -0.2270+--0.1080
l: 0.60 (w=0.100) cont: -0.0850+--0.0710
l: 0.70 (w=0.100) cont: -0.0030+-0.0120
l: 0.80 (w=0.100) cont: 0.0500+-0.1030
l: 0.90 (w=0.100) cont: -0.0300+-0.0070
l: 1.00 (w=0.050) cont: -0.0550+-0.0035
Total DV/DL:-0.8995+--0.242
Step3
l: 0.00 (w=0.050) cont: -0.0135+--0.0060
l: 0.10 (w=0.100) cont: -0.0300+--0.0120
l: 0.20 (w=0.100) cont: -0.0330+--0.0120
l: 0.30 (w=0.100) cont: -0.0200+--0.0120
l: 0.40 (w=0.100) cont: -0.0040+--0.0120
l: 0.50 (w=0.100) cont: -0.0070+--0.0130
l: 0.60 (w=0.100) cont: -0.0170+--0.0120
l: 0.70 (w=0.100) cont: -0.0260+--0.0120
l: 0.80 (w=0.100) cont: -0.0190+--0.0130
l: 0.90 (w=0.100) cont: -0.0190+--0.0120
l: 1.00 (w=0.050) cont: -0.0095+--0.0060
Total DV/DL:-0.198+--0.122
Total free energy diff is -1.1085
Any help you could provide would be greatly appreciated. I have enclosed graphs of DV/DL vs lambda for the three stages of the complex.
G. Tampi
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
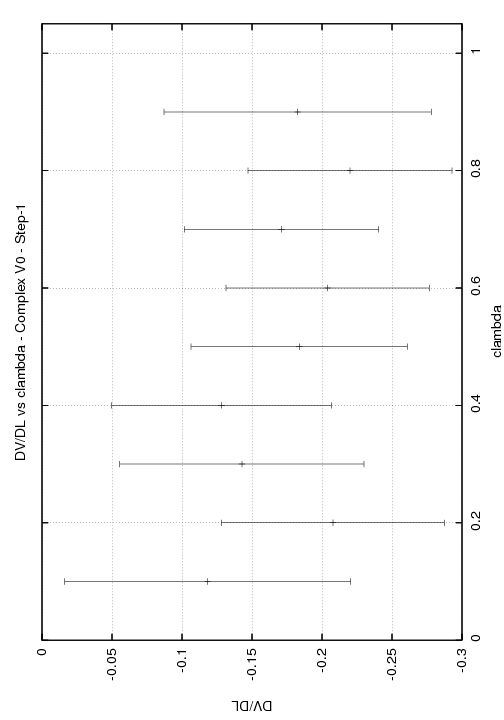
(image/jpeg attachment: DV_DL_step1_V0_complex.jpg)
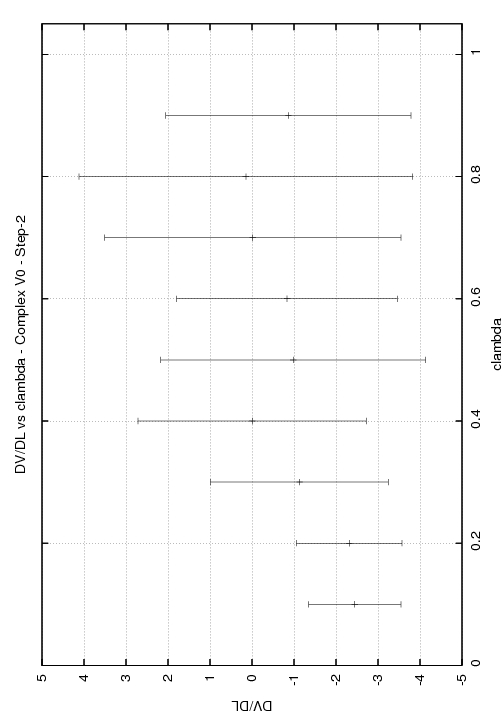
(image/jpeg attachment: DV_DL_step2_V0_complex.jpg)
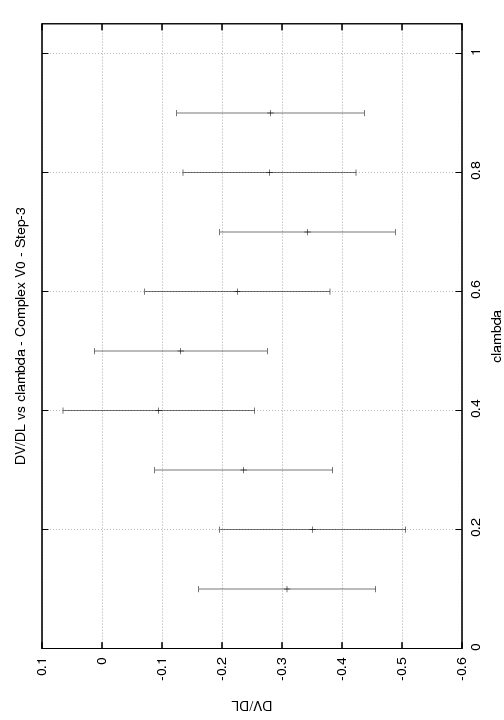
(image/jpeg attachment: DV_DL_step3_V0_complex.jpg)
