Dear Jiomm,
I can see where the confusion is... the structure in the article does not
give any indication as to the bonding in the structure. For structures like
yours, a pdb is the wrong way to go because asking any app to guess the
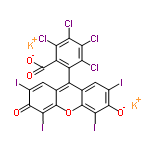
bonding types is virtually impossible. All the rings in your system are
aromatic, with O atom in the aromatic ring, yet no Lewis formula can
correctly draw that, (as evidenced by the first two structures I've provided
you that I found with a quick google search using "bengal rose".)
You also need to question why the article used the protonated carboxyl and
protonated phenolic form.
I have provided for you the pKa predictions for the phenolic and carboxylic
hydrogens for this compound: the carboxyl pKa is +0.61 and the phenolic pKa
is +3.33. From the titration curve I provided you, it is clear that the
carboxyl is about 99% deprotonated before the phenolic group even begins to
deprotonate.
The structure as shown in the article with a protonated carboxyl and
protonated phenol group (neutral charge) would be the predominant species
only at a pH of -2.61 or lower !!! At pH 2.0 compound would be 92% in the
carboxyl deprotonated, phenolic protonated form (-1 charge) and anything
above pH 5.33 would virtually 100% deprotonated carboxyl and phenol (-2
charge). You need to seriously consider the pH at which you want to
simulate, because that will completely determine which of the three
protonation forms you need to parameterize.
Assuming your work is done at anything near physiologic conditions, then
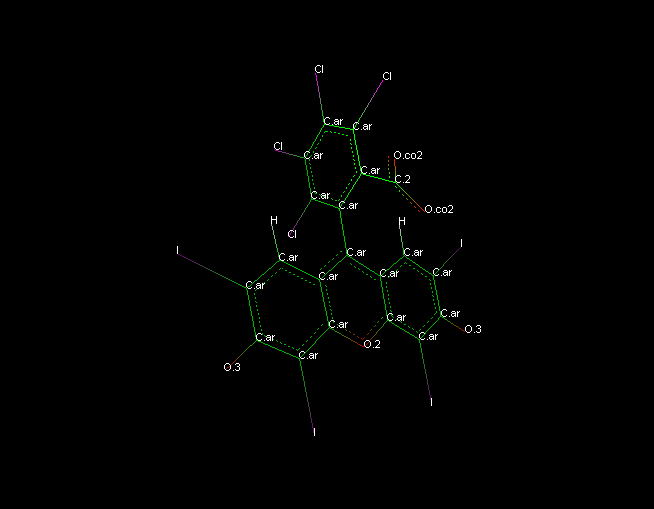
image sybylatomtypes.mol2.png shows your -2 charge structure with correct
hybrid resonance and appropriate sybyl atom types. Note that some of these
atom types I had to fix by hand, because the chem structure program (VEGA ZZ
2.3.1) did not identify all the carbons in the 3 ring system as being
aromatic. Also note that the three ring system is completely symmetric
(except for some asymmetry of charge due to the carboxyl plane being
skewed), so both oxygens should equally share the negative charge and
therefore be equal and very close to -0.5
Using this mol2 file as input to antechamber gave me a new mol2 file with
gaff atom types. These were close to correct, but still needed manual
correction to make the three ring system carbons type ca except for the one
connecting by single bond to the other aromatic ring, where the correct type
would be cp.
I also gave you the antechamber and parmchk output to use. The only
troubling atom type is the oxygen in the central aromatic ring. This would
need to be modified, and it has been already parameterized as previously
discussed on this list, since gaff does not have an atom type for an
aromatic oxygen.
I hope this helps, and I too had to remind myself that Lewis structures do
NOT work many of the times, and that the truth of the structure lies beyond
a single Lewis representation, and of course the Huckel rule (4N + 2) :-)
Good luck with your work,
Dean
Dr. Dean Cuebas dac012f.missouristate.edu,
Ph 417-836-8567 FAX 417-836-5507
Dept. of Chemistry, Missouri State University
Springfield, Missouri 65897
> From: Jio M <jiomm.yahoo.com>
> Reply-To: AMBER Mailing List <amber.ambermd.org>
> Date: Thu, 10 Dec 2009 05:03:59 -0600
> To: AMBER Mailing List <amber.ambermd.org>
> Subject: Re: [AMBER] Re: COOH troubling
>
> Dear Dr. Dean and J. Wang;
>
>
>
>> Either get rid of the double bond and add a negative charge to make a
>
> phenolate ion, or make a C-O-H by adding a hydrogen to the oxygen to make a
>
> phenolic group, like you have on the opposite side of your anthracene ring
>
> system.
>
>
>
> I am sending link which shows my structure.
>
> http://www.wag.caltech.edu/publications/sup/pdf/370.pdf
>
> But I have used RESP charges. There some other charge method is used. I think
> it is C double bond O.
>
>
>
> thanks and regards;
>
> JIomm
>
>
>
>
>
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Thu Dec 10 2009 - 23:00:02 PST
{kind=link}
{kind=link}
