Dear Sir,
I'm very sorry not to send enough information to you.
At first, I tried to make a file by using xleap following the tutorial B1.
Only one changed point is a solute from DNA to a protein.
The command files for MD are the same of the tutorial B1.
>We need more information (but probably not all your files): what is the
>*exact* symptom of the problem, that is: what is it makes you conclude that
>some water atoms were recognized as the atoms of the protein?
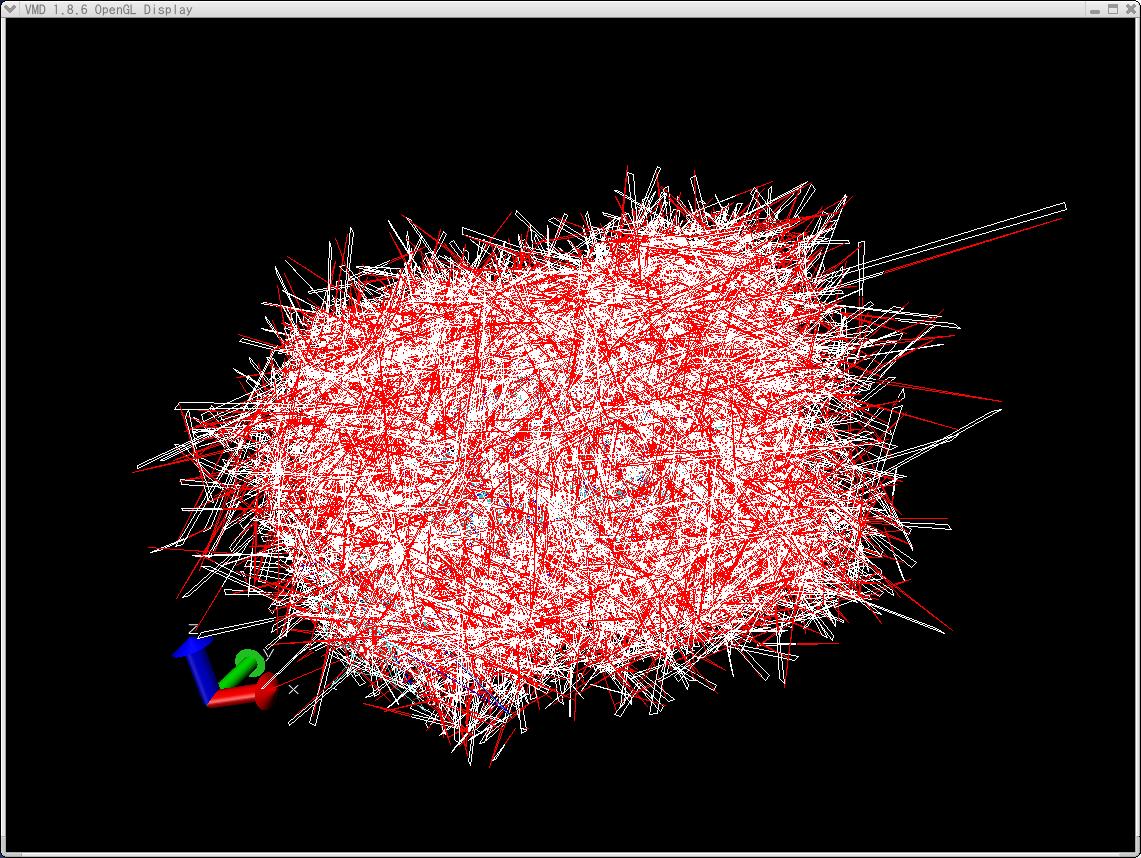
I check the resulted trajectory files by using VMD. Some very long bonds
assigned to protein (the color is gray and blue) could be seen between the
outside and the inside of the protein.
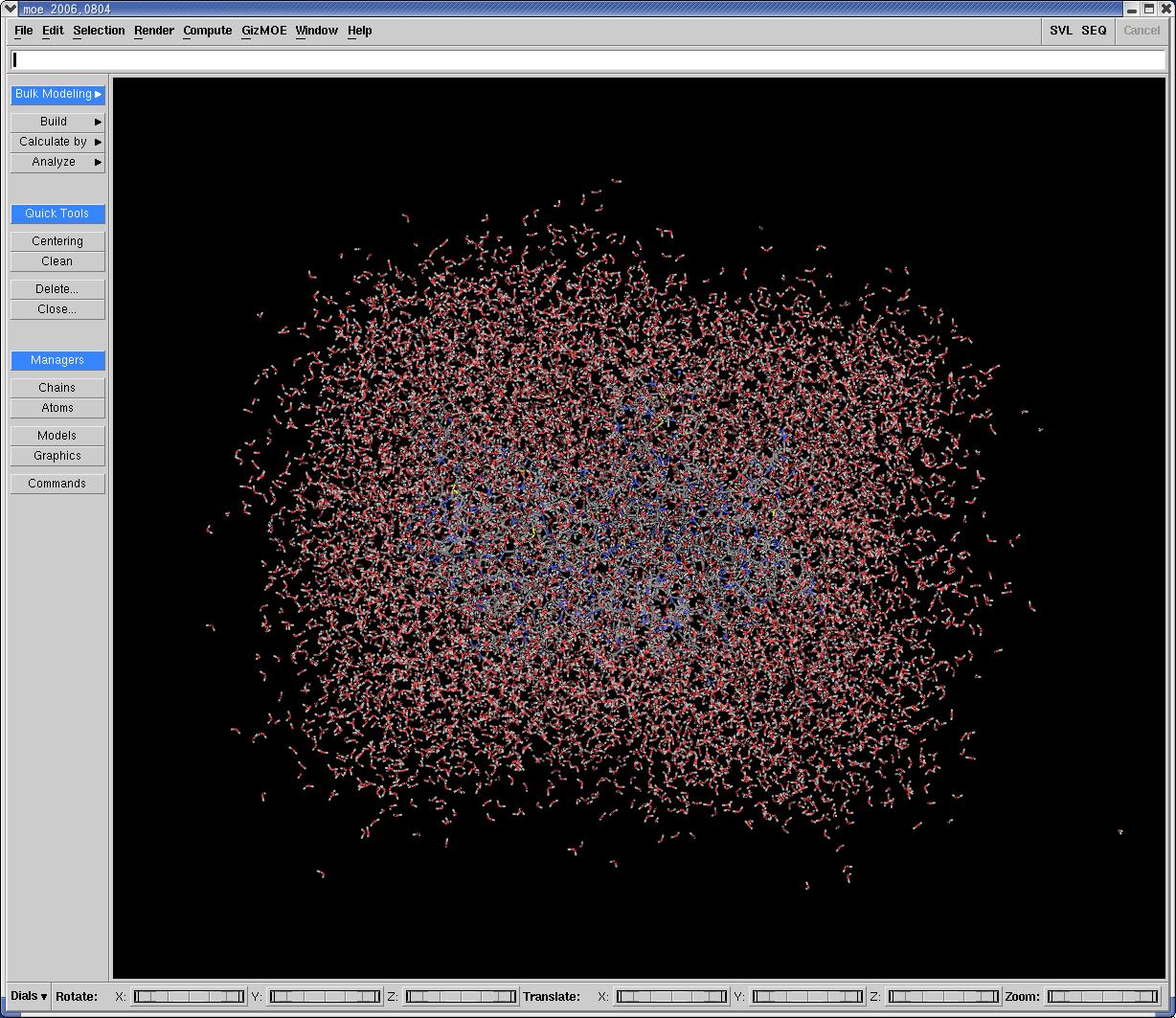
I made PDB files from the trajectory file by using ptraj every 1ps.
I visualized some PDB files in the MOE (Molecular Operating Environment).
Some long bonds could be seen and the situation of the some points were
seemed to be that of water molecular.
I wonder where is wrong for this calculation.
>What commands did you give to LEaP to prepare the explicit solvent system?
When I use the "test.pdb" file as the input, I used the following commands.
pp=loadpdb "test.pdb"
addions Na+ 0
saveamberparm test.prmtop test.inpcrd
That's all.
I think one reason may be derived from VMD.
The attached file, named b1_md2_189.jpg, shows a snapshot of MD.
The file, named b1_md2_moe.jpg, shows the same snapshot visualized by MOE
(translated to PDB format by ptraj). The b1_md2_181.jpg file is the other
snapshot figure made by the same procedure.
The figure by VMD was something wrong.
>Did you do something different that what is in tutorial B1?
Next, I made some changes in the input file of MD.
I compiled AMBER on the several platform. The results of one cmoputer seem
good but another computer does not work. It may be compile error. Could I
ask you about compile errors later.
Sincerely,
Miki
PS
I wonder whether I send it to the mailing-list because big files may make
a trable with each reader of the mailer/mailbox. I'm grad if I can directly
submit them.
_/_/_/ M. MAEDA mmaeda.nias.affrc.go.jp _/_/_/
_/_/_/ National Institute of Agrobiological Sciences _/_/_/
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber.scripps.edu
To unsubscribe, send "unsubscribe amber" to majordomo.scripps.edu
Received on Sun Jul 15 2007 - 06:07:18 PDT
{kind=link}
{kind=link}
{kind=link}
